La secuenciación masiva permite secuenciar el genoma humano por un precio alrededor de 1000 dólares. Dicha secuenciación a una cubertura de 30 por (cada posición se secuencia 30 veces de media) produciría alrededor de 600 millones lecturas de una longitud de 150 nucleótidos. Para poder detectar variación de secuencia en la muestra secuenciada (el genoma de un individuo) hay que alinear todas las lecturas a un genoma de referencia (la secuencia consenso de un grupo de personas que representa el genoma de una población o especie). Las lecturas apiladas en una posición nos pueden indicar:
- si hay variación de una base y el genotipo correspondiente
- si existen inserciones o deleciones cortas
La detección de la variación de secuencia tiene muchas aplicaciones:
- medicina personalizada: relacionar la existencia de ciertas variantes con el pronóstico de una enfermedad o la individualización de la terapia
- diagnóstico: detectar la predisposición a ciertas enfermedades
- pronóstico: predecir el transcurso de una enfermedad
- hacer pruebas forenses o de paternidad
En el genotipado masivo la bioinformática tiene un papel importante. La Ilustración abajo muestra las lecturas de un individuo alineadas frente al genoma de referencia. Mediante estos datos, aplicando métodos estadísticos se puede determinar el genotipo para cada posición del genoma.
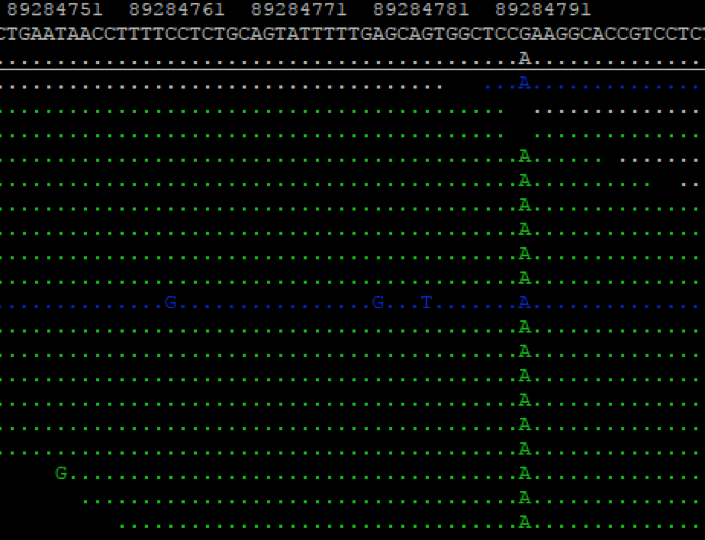
Apilamiento de lecturas obtenidos mediante secuenciación masiva del genoma de un individuo. Se puede observar que este individuo en la posición 89284793 del cromosoma 11, es homocigoto para el alelo A siendo ‘G’ el alelo de la referencia del SNP rs1126809. Este genotipo está relacionado con Albinismo al cambiar la actividad enzimática de la tirosinasa
Por el otro lado, la secuenciación masiva genera muchos datos, pero la ratio entre señal y ruido no es muy alta. Por ejemplo, si secuenciamos el genoma de un individuo y analizamos los nucleótidos en cada posición, veremos que frecuentemente no solamente encontramos el nucleótido de la referencia sino también otros a frecuencias más bajas. Si encontramos un segundo nucleótido a frecuencias cercanas al 50% podemos concluir que el individuo es heterocigoto, pero frecuentemente nos encontramos con situaciones como la mostrada en la Ilustración abajo. En esta posición hay una adenina en el genoma de referencia y el individuo muestra adenina en 9 de 11 lecturas que alinean frente a este locus. En dos lecturas hay una timina y el gran reto es saber si se debe a errores de secuenciación y/o de la PCR, si es otro alelo (el individuo seria heterocigoto A/T) o si es una mutación somática y por eso no está presente en todas las células que se han usado para la secuenciación (y por eso la frecuencia del alelo es mucho más baja).
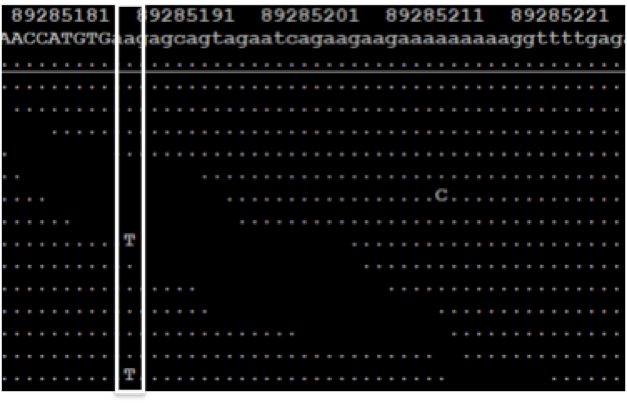
Una posición que muestra variación A/T y que puede deberse a diferentes razones: errores de secuenciación, variantes de secuencia o mutaciones somáticas