En esta práctica vamos a genotipar parejas en las que al menos uno de los dos tiene antecedentes familiares para alguna enfermedad heredable (enfermedad mendeliana). Para determiniar el genotipo de los individuos disponemos de datos de secuenciación masiva.
Datos: 29-A_read1.fq
Para genotipar tenemos que: i) alinear las lecturas, ii) convertir el formato, iii) apilar las lecturas, iv) determinar si hay variación y determinar el genotipo
0) indexar la secuencia de referencia (mediante sametools faidx)
samtools faidx <reference.fa>
Este paso ya se llevó a cabo para la secuencia del genoma humano hg38.fa que usaremos en esta práctica. El resultado de este comando es un nuevo fichero con el mismo nombre base que la entrada y extensión .fai. Ese fichero es el índice de la referencia en fasta.
1) alinear las lecturas
bowtie2 -x /opt/genomes/hg38 -U 29-A_read1.fq_.gz > 29-A_read1.sam
Formato sam: https://genome.sph.umich.edu/wiki/SAM
El formato sam contiene la información de las lecturas ya alineadas. Es un fichero de texto plano (podéis abrirlo con cualquier editor y comprobar que se tratan de filas tabuladas). El archivo binario equivalente es bam, y contiene la misma información solo que almacenada de forma binaria. Esto hace que muchos programas puedan interactuar más eficientemente. De hecho, muchos programas requieren que el fichero de lecturas alineadas esté ya en formato .bam (y a veces también se requiere que esté ordenado, i.e. primero por cromosoma y después por coordenadas cromosómicas).
2) convertir el formato SAM en BAM
samtools view -S 29-A_read1.sam -b -o 29-A_read1.bam
3) ordenar el bam (por coordenadas)
samtools sort 29-A_read1.bam > 29-A_read1_sort.bam
Una vez que hayamos obtenidos un fichero bam ordenado, podemos llevar a cabo dos tipos de análisis: i) visualizar el apilamento de lecturas y ii) detectar la variación y el genotipo
Visualizar
4) determinar la primera base de la región con alineamientos
samtools view 29-A_read1_sort.bam | more
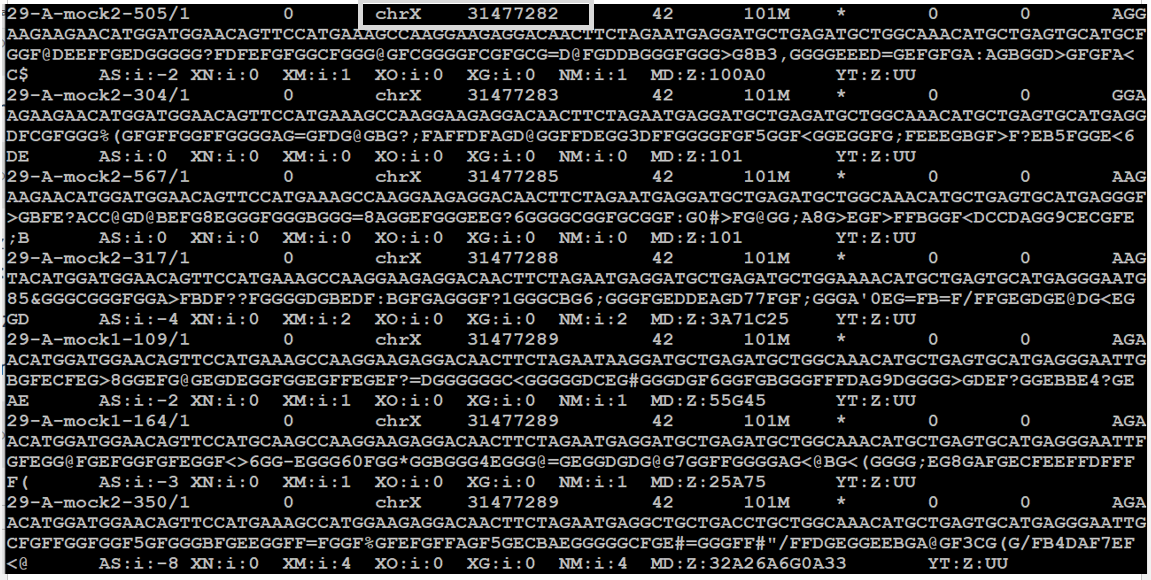
Fijándonos en la primera línea del fichero (desde 29-A-mock2-505/1 hasta la siguiente línea que comienza casi igual), podemos ver que el primer campo es el identificador de la lectura, el segundo es la flag (0, que quiere decir que es una lectura mapeada) el siguiente corresponde al cromosoma y el siguiente a la posición cromosómica de inicio del alineamiento.
5) generar índice del fichero BAM (.bai)
samtools index 29-A_read1_sort.bam
6) visualizar los alineamientos apilados
samtools tview 29-A_read1_sort.bam /opt/genomes/hg38.fa -p chrX:31477282
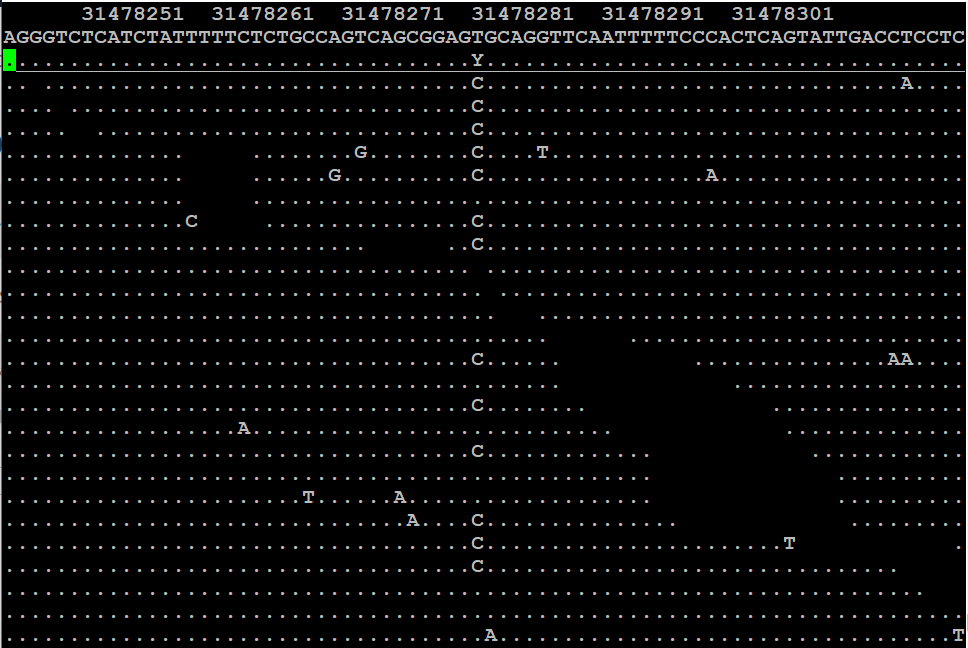
Podéis navegar usando las teclas de flecha. En la primera línea podéis ver la coordenada cromosómica y en la segunda la secuencia de referencia. Cada fila debajo de la línea continua se corresponde a un read alineado. Un punto indica que la secuencia del read corresponde a la referencia y una letra indica un nucleótido alternativo a la referencia para ese read concreto.
Detectar la variación y el genotipo
Como podéis ver en el fichero, hay distintos reads que difieren respecto a la referencia en (al menos) una posición. ¿Quiere esto decir que se trata de una variante? Las diferencias resaltan más que las similitudes pero recuerda que todos los puntos quieren decir que esa posición concuerda con la referencia. Por lo tanto, si en una posición mapean 10 reads y solo uno contiene un nucleótido diferente a la referencia, ¿debemos pensar que se trata de una variante? La secuenciación es un proceso con relativa alta precisión pero si cada read tiene longitud 100, asumiendo una precisión del 99%, esperamos (de media) un nucleótido “erróneo” por cada read.
Estos factores se han tenido en cuenta a la hora de desarrollar los programas que permiten determinar el genotipo (variant calling).
7) Apilar las lecturas
//samtools mpileup [options] in1.bam [in2.bam [...]] samtools mpileup -g -f /opt/genomes/hg38.fa 29-A_read1_sort.bam > 29-A_read1_sort_raw.bcf -g --> genera salida bcf (formato binario del vcf, variant call format)
8) Determinar la variación y el genotipo
bcftools call -c -v 29-A_read1_sort_raw.bcf > 29-A_read1.vcf // -c --> SNP calling // -v --> only variant sites
descarga el fichero y ábrelo en un editor de texto.
resultado: últimas 2 lineas del fichero 29-A_read1.vcf (el resto es el encabezado)
Explicación detallada del formato vcf
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 29-A_read1_sort.bam
chrX 31478281 . T C 130 . DP=33;VDB=1.260432e-01;RPB=-2.141714
e+00;AF1=0.5;AC1=1;DP4=13,0,18,0;MQ=42;FQ=119;PV4=1,0.17,1,0.46 GT:PL:GQ 0/1:160,0,146:99
Campos importantes
- chrX 31478281: cromosoma y posición
- DP: el número de lecturas mapeadas
- AF1: frecuencia alélica del primer alelo alternativo
- GT: el genotipo, 0/1 significa que es heterocigoto alelo de referencia (T) & primer alelo alternativo (C)
9) Determinar si se conoce la variación
Hasta aquí hemos completado la parte puramente técnica del ejercicio: a partir de los datos de secuenciación hemos determinado el genotipo del individuo. En los siguientes pasos intentaremos averiguar varias cosas:
- si esta variante es conocida
- si se encuentra en algún lugar del genoma y si puede corresponder a un cambio en la traducción de una proteína
- consecuencias fenotípicas de ese posible cambio
usa el navegador del UCSC para ver si hay un SNP anotado en esa posición. Introduce la posición y navega al track dbSNP (actívalo si no lo está). Pinchando en el track te llevará a un panel donde aparece el identificador del SNP (típicamente empiezan por rs)
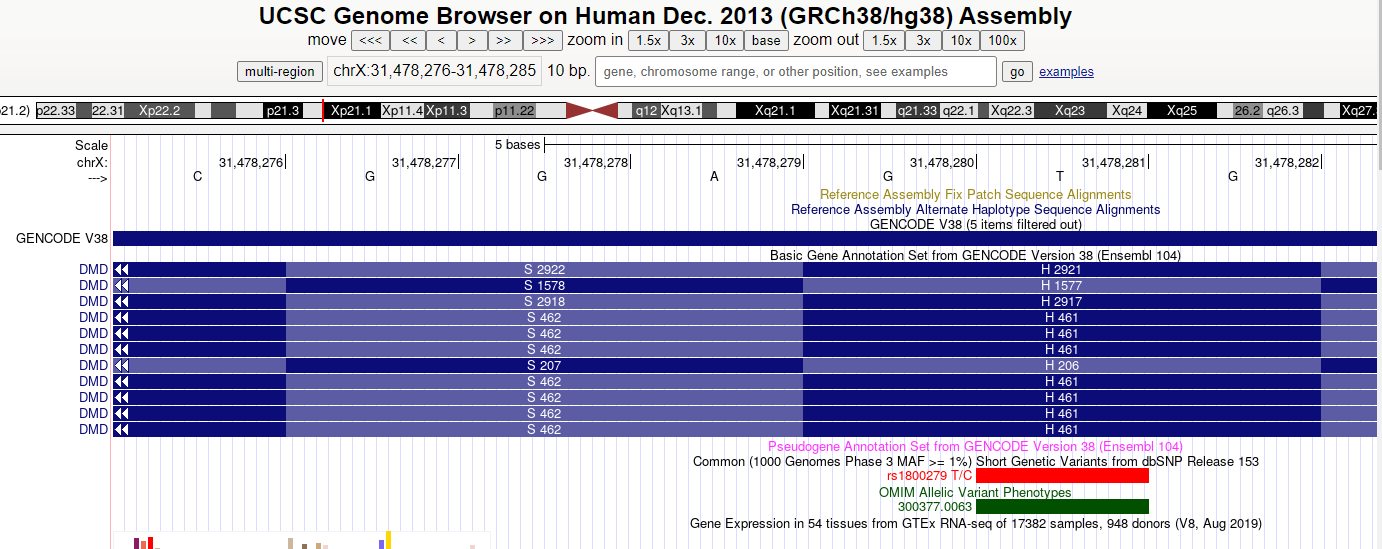
10) Determinar el efecto usando la dbSNP
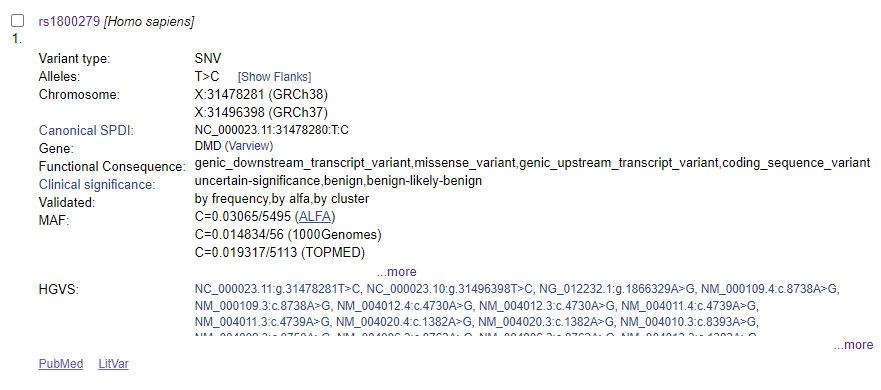
La posición chrX:31478281 corresponde al SNP rs1800279.
si lo buscamos en la base de datos dbSNP encontramos que causa un cambio de sentido erróneo (missense) CAC ⇒ CGC en diferentes transcritos del gen DMD
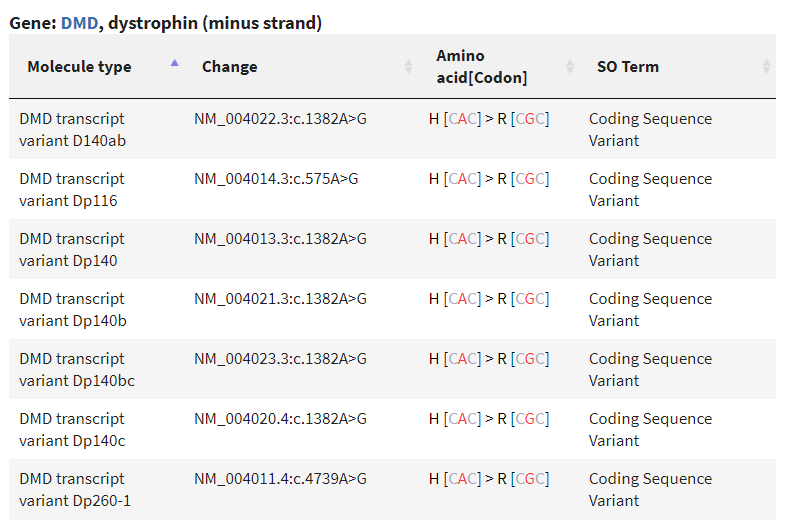
Importante
el gen DMD se ubica en la hebra ‘-‘ y por eso se dan los dos alelos como A y G, y no como T y C detectados arriba en la hebra ‘+’
11) buscar el fenotipo asociado las bases de datos ClinVar y OMIM
Puesto que defectos en el gen DMD están asociados a una distrofia muscular, debemos comprobar en estas dos bases de datos si se han descrito casos donde esta variante sea responsable de la enfermedad.
Comprobando la información disponible en ambas bases de datos, podemos concluir que esta variante no parece tener efectos patogénicos a pesar de alterar la secuencia de aminoácidos de la proteína resultante (Likely benign/ benign).
Cuestiones*:
- ¿Cual es la frecuencia del alelo menor?
- Si este individuo (mujer) tuviera un hijo con el individuo 29-C (29-C_read1.fq), ¿cual sería la probabilidad de que sus hijos/hijas padezcan la distrofia muscular?
- Qua es la probabilidad de que los niños/niñas de los siguientes individuos padezcan distrofia muscular: Padre: 29-D_read1.fq ; Madre: 29-B_read1.fq
Recomendaciones y más información:
- Leer este capítulo: http://www.sciencedirect.com/science/article/pii/B9780124047488000083
- Si ninguna variación de secuencia es detectada, podemos cambiar algunos parámetros en el paso del mpileup para hacer la detección más laxa: http://www.htslib.org/doc/samtools.html
-B: deshabilitar el realineamiento probabilistico
-E: recalculando la calidad del alineamiento
-Q: cambiar la calidad mínima de una base
3. Calcular la probabilidad de que exista variación de secuencia mediante una tabla de contingencia: http://vassarstats.net/tab2x2.html
*Nota del profesor: este ejercicio se preparó cuando la evidencia disponible apuntaba a que esta variante SÍ PROVOCABA Distrofia muscular de Duchenne. Para realizar el ejercicio propuesto, considera que esta variante SÍ PROVOCA la enfermedad (a pesar de que los datos actualmente indican que no).