La densidad génica se refiere al número de genes presentes en una región específica del genoma y varía considerablemente entre especies. En organismos con genomas compactos, como las bacterias, la densidad génica suele ser alta, con pocos o ningún espacio entre genes, mientras que en organismos más complejos, como los mamíferos, los genes suelen estar separados por grandes regiones de ADN no codificante. El estudio de la densidad génica es fundamental para entender cómo las diferentes especies organizan y regulan su material genético. Esta variabilidad influye en procesos biológicos como la replicación, transcripción y evolución del genoma. Además, analizar las diferencias en la densidad génica entre especies permite desentrañar las adaptaciones evolutivas y comprender mejor la relación entre la complejidad del organismo y la organización de su genoma.

En estas prácticas calcularemos la densidad génica de dos maneras diferentes:
- Porcentaje de bases codificantes: (nucleótidos en CDS / número totales de nucleótidos) * 100
- Número de genes por Mbp: (número de genes * 1.000.000) / número totales de nucleótidos
Porcentaje de bases codificantes
Para determinar el porcentaje de bases codificantes, primero vamos a añadir los datos de una tabla de genes al historial de Galaxy.
En el menú Get Data seleccionaremos UCSC mainRecordad: Si usamos genomas de GenArk, debemos cargarlos previamente en el UCSC
Galaxy nos redirigirá al la herramienta table browser de la UCSC, además de la especie y el ensamblado, seleccionaremos Genes and Gene Predictions, NCBI RefSeq y nos quedaremos con la tabla all. Debemos recordar filtrarlo por el cromosoma que estemos analizando y el formato de salida será BED.
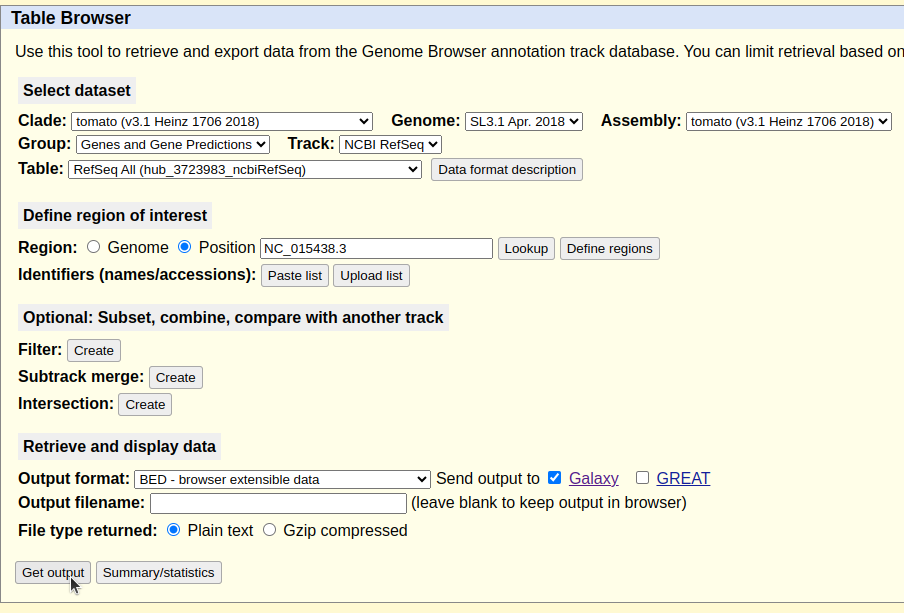
En la siguiente ventana se nos permitirá seleccionar qué regiones de los genes queremos obtener, en este caso buscamos las regiones codificantes (CDS).
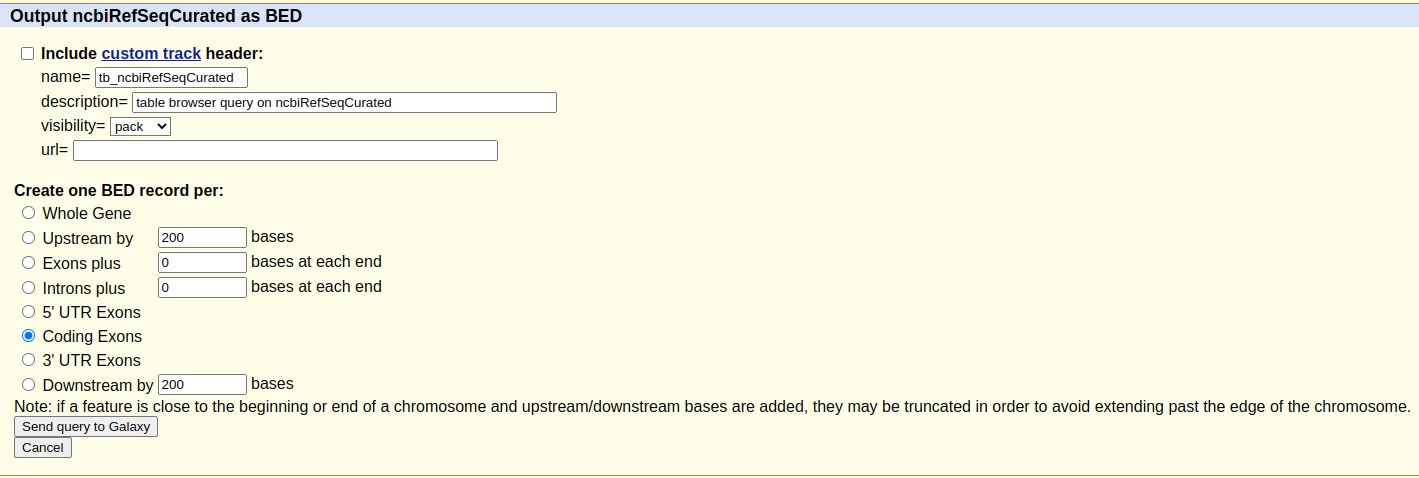
Este proceso añadirá un nuevo objeto a nuestro historial en Galaxy que incluirá todos los CDS del cromosoma seleccionado.
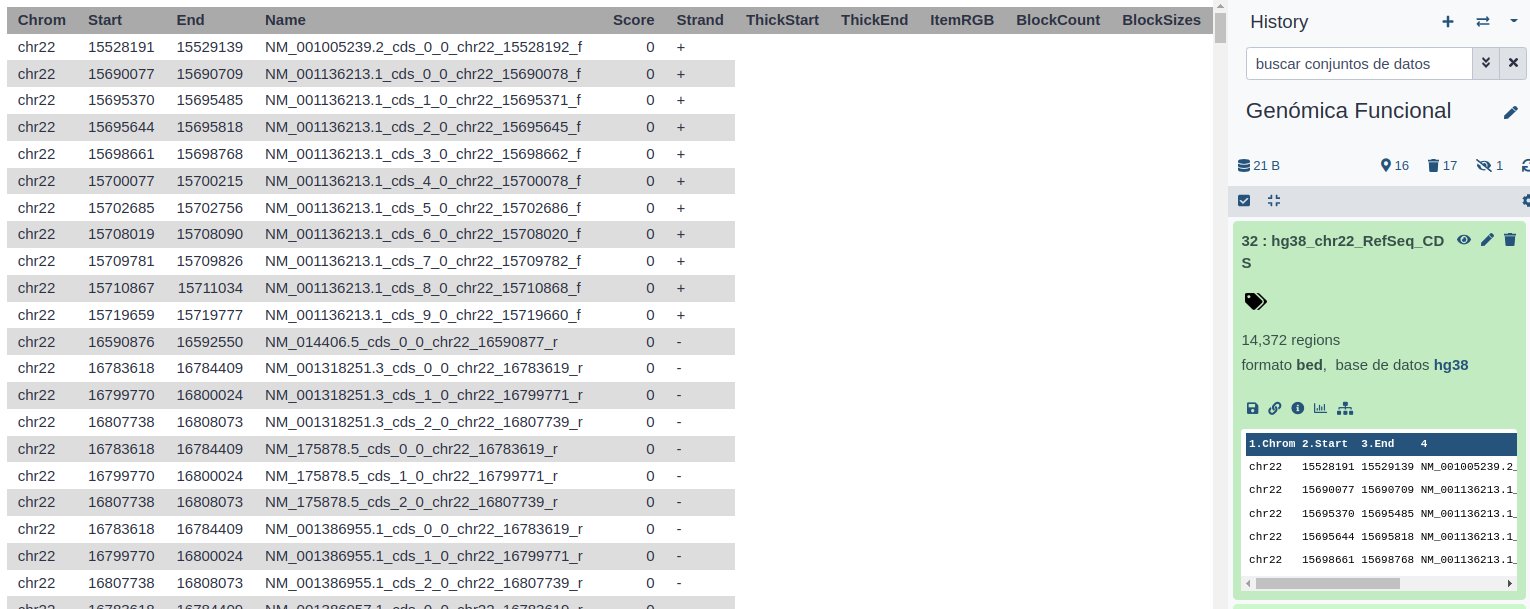
Ahora para calcular el porcentaje de bases codificantes debemos calcular el número de bases que ocupan y dividirlo por la longitud total de la secuencia (sin Ns). En primer lugar, para calcular el número de bases que codifican proteínas, debemos tener en cuenta que la mayoría de los genes tienen más de un transcrito anotado, y estos comparten exones. Por lo tanto, tenemos que eliminar esta redundancia, para no sobreestimar el % del genoma que codifica proteínas.
Utilizaremos la función Merge del menú Operate on Genomic Intervals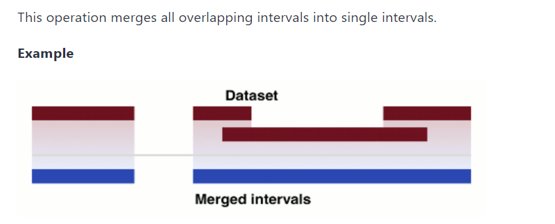
Seleccionaremos el fichero de CDS y la opción Output 3 column bed.
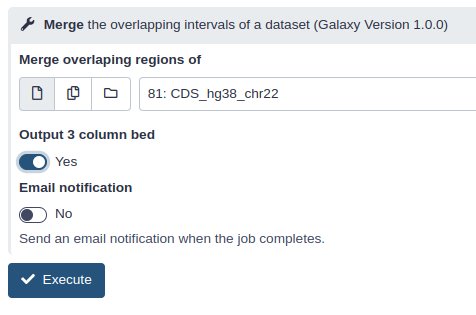
Esto nos devolverá todas las regiones de CDS no solapentes del cromosomas con las que calcularemos la suma total de nucleótidos que se encuentran en CDS.
Seleccionamos la función Summary Statistics del menú Statistics
- Calculamos la estadística de las longitudes de las regiones (Final - Inicio)
La función nos devolverá la suma de los valores, la media, la desviación estándar y los cuartiles de valores de la distribución. Con esto, la información obtenida del cromosoma completo y la formula enunciada arriba, ya podemos calcular la densidad de secuencias codificantes de nuestros crosomas.
ANTES DE CONTINUAR COMPLETA EN LA TABLA COMPARTIDA:
La densidad de secuencias codificantes
La densidad de islas CpG
Número de genes por megabase
La medida de genes por Mbp es la más utilizada en los estudios comparativos, pero para calcularla correctamente a partir de las anotaciones debemos tener en cuenta que un mismo gen presenta múltiples isoformas, por lo que cada gen deberá contabilizarse de manera única evitando así sesgar el recuento por el número de isoformas anotadas. Además, las anotaciones de CDS no nos sirven para este propósito por lo que deberemos descargar la información génica en un formato diferente. Realizaremos los mismos pasos que para cargar los CDS, pero modificaremos la salida como se observa en la imagen inferior.

Y en la siguiente pestaña seleccionaremos los siguientes campos.

El objeto resultante contendrá 7 columnas que incluyen:
- name: nombre de la isoforma anotada.
- chrom: nombre del cromosoma donde se localiza.
- strand: hebra en la que se transcribe.
- txStart/chromStart: sitio de inicio/final de la transcripción dependiendo de si se transcribe en la hebra +/- respectivamente.
- txEnd/chromEnd: sitio de inicio/final de la transcripción dependiendo de si se transcribe en la hebra -/+ respectivamente.
- exonCount/blockCount: número de exones anotados en la isoforma.
- name2: nombre del gen al que pertenece la isoforma.
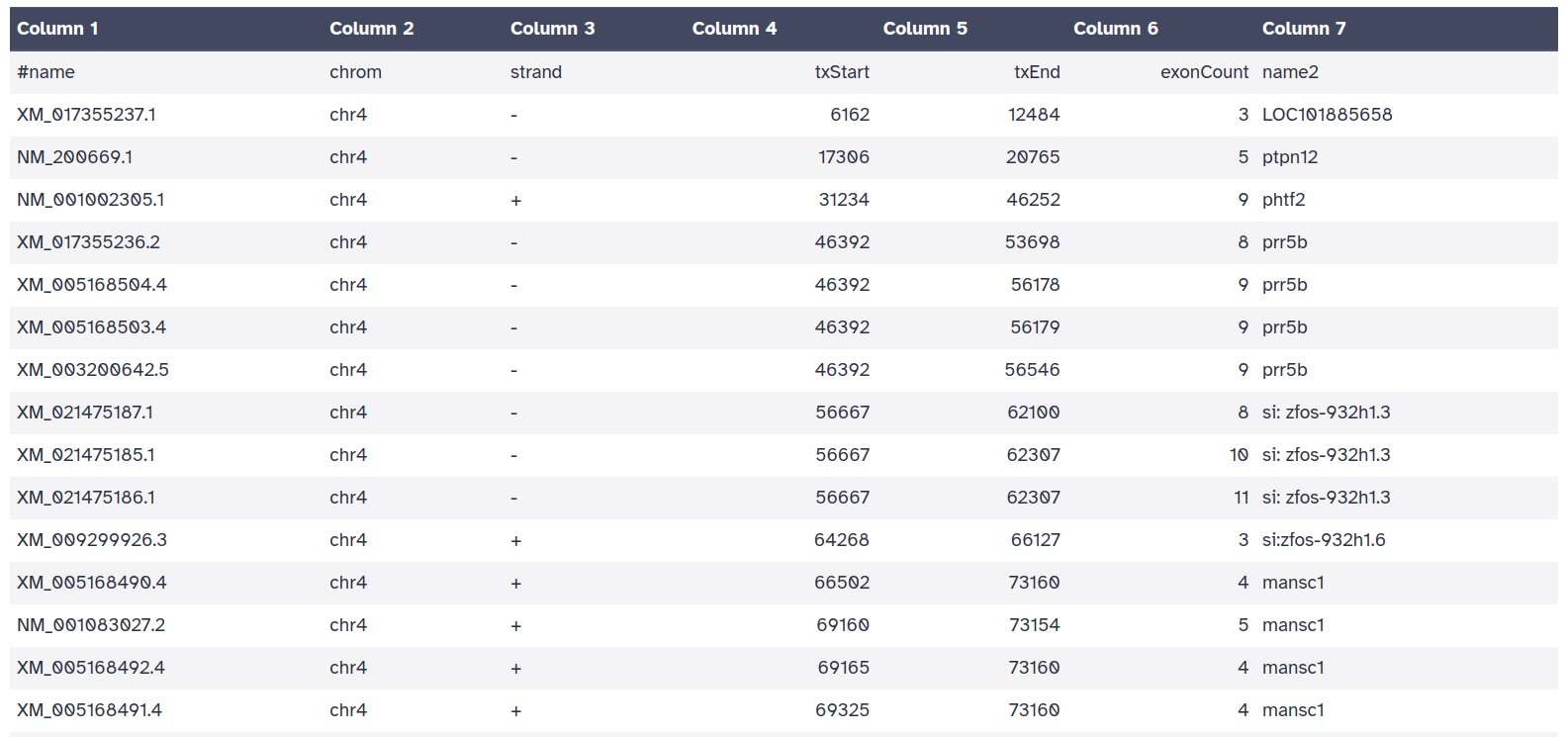
Por lo tanto, para obtener el número de genes únicos en el cromosoma deberemos eliminar la redundancia de la columna name2.
Utilizaremos la herramienta Count del menú Statistics
- En la opción Count ocurrences of values in column(s) seleccionaremos name2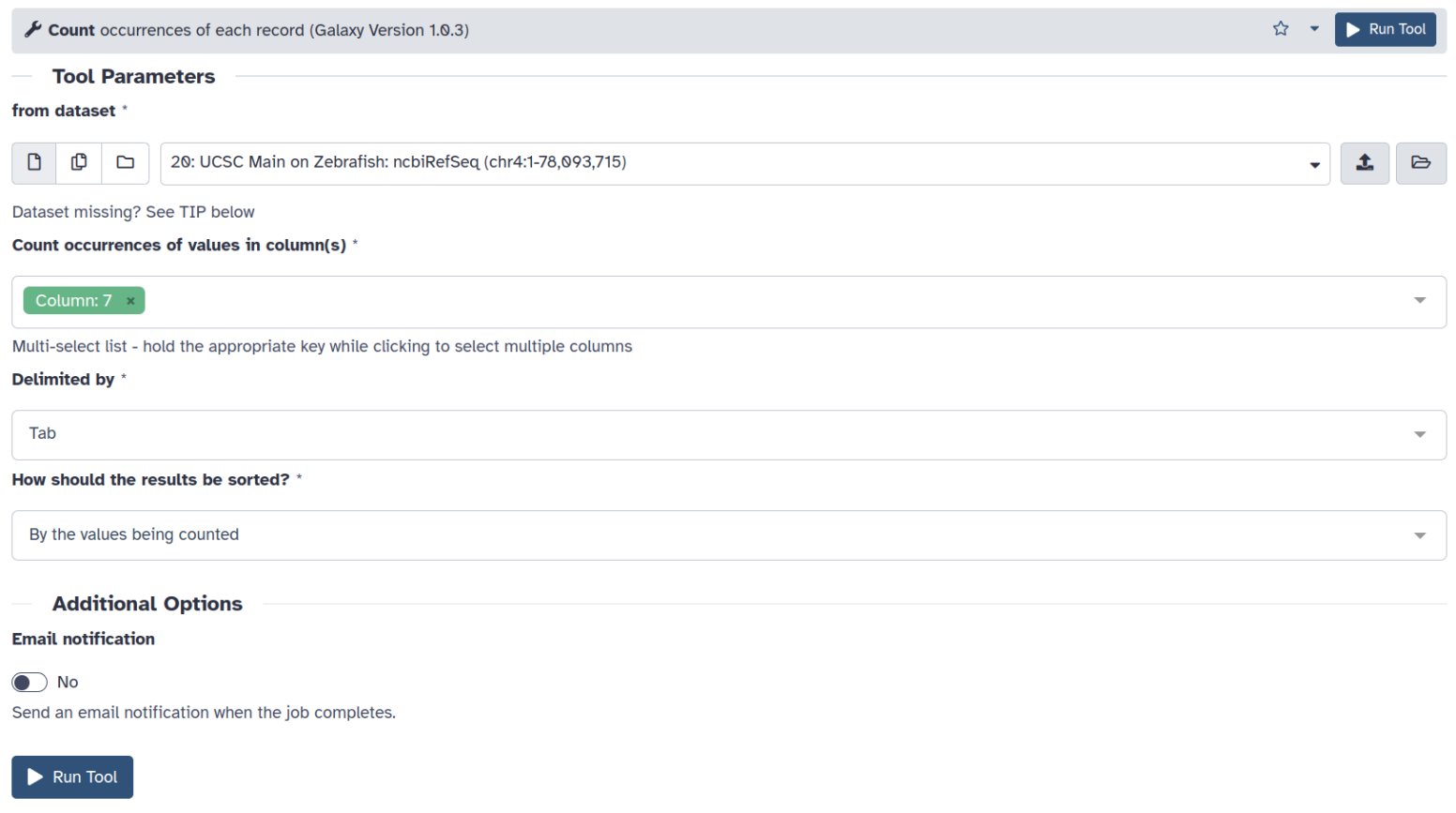
De esta manera obtendremos el número de isoformas para cada gen en name2, representando el número de líneas del nuevo objeto el número de genes únicos presentes en el cromosoma.
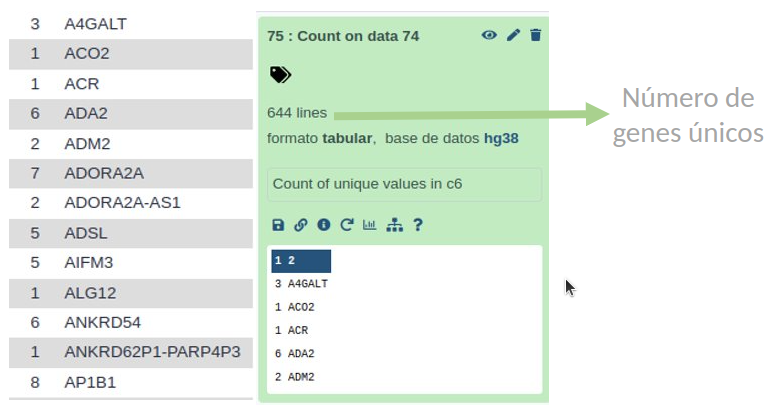
ANTES DE CONTINUAR COMPLETA EN LA TABLA COMPARTIDA:
El número de genes
Filtrar tablas en base a patrones de texto
La gran mayoría de las veces no queremos cuantificar la anotación completa que descargamos de las bases de datos por lo que tenemos que preseleccionar algunos de los elementos en base a la información recogida en las tablas. Con este fin podemos utilizar la identificación de patrones de texto para filtrar las tablas antes de cuantificar nuestros genes o elementos genómicos.
En el caso de genes, la mayoría de las veces nos interesa analizar genes codificantes y no-codificantes de manera separada. Por suerte, el propio identificador de las isoformas nos puede permitir separarlos, ya que las letras al inicio de los identificadores de RefSeq hacen referencia al tipo de isoforma de que se trata como puede apreciarse en la tabla siguiente.
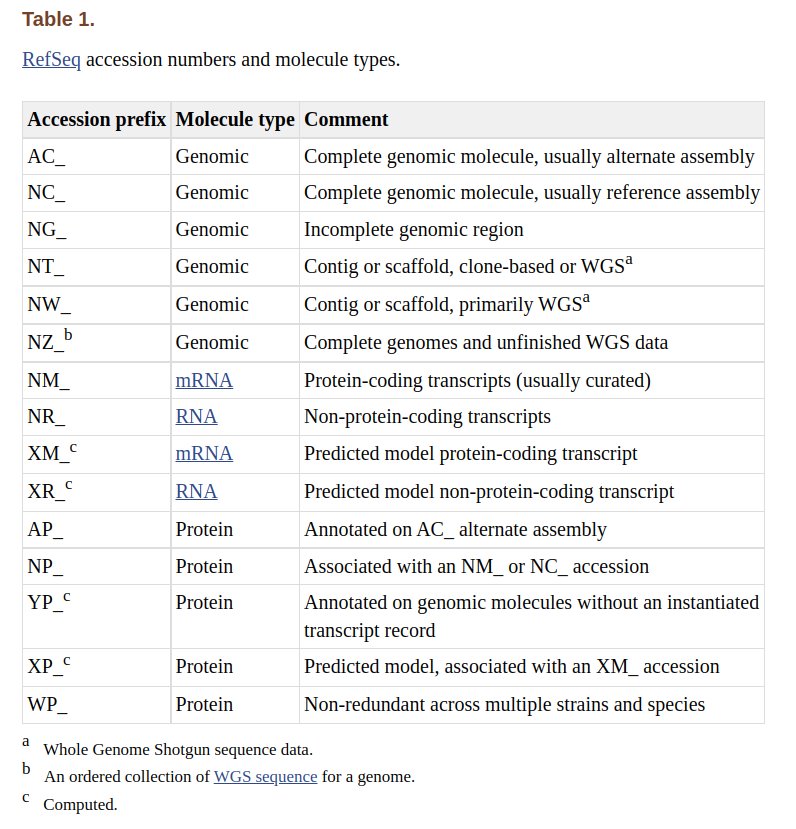
La herramienta Select del menú Filter & Sort nos permite seleccionar aquellas líneas que presenten un patrón de textoRESUELVE ANTES DE CONTINUAR:
Diseña un flujo de trabajo que permita calcular la densidad de genes codificantes y no-codificantes por Mbp
Diseña un flujo de trabajo que permita calcular el porcentaje de nucleótidos de genes codificantes y no-codificantes
Completa los datos en la tabla compartida
PISTAS
- El patrón para genes codificantes es NM_|XM_
- El patrón para genes no-codificantes es NR_|XR_
- Consideramos que los nucleótidos en un gen son aquellos entre el inicio y el final de su transcripción
- Recordad que puede haber redundancias entre isoformas que deben evitarse
- En algunos casos, los elementos de la tabla descargada pueden estar en un orden no compatible. Para reordenar las columnas deberemos hacer uso de la herramienta cut.