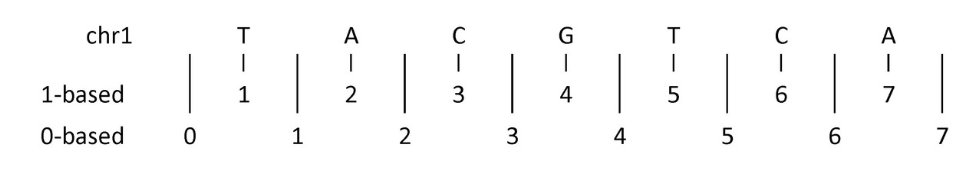
Especies a Analizar
| Especie | Ensamblado | Base de Datos |
|---|---|---|
| Human | hg38 | UCSC |
| Mouse | mm39 | UCSC |
| Cat | felCat9 | UCSC |
| Cow | bosTau9 | UCSC |
| Chicken | galGal6 | UCSC |
| Zebra finch | taeGut2 | UCSC |
| Turkey | melGal5 | UCSC |
| American alligator | rAllMis1 | GenArk |
| Green anole | rAnoCar3.1.pri | GenArk |
| Corn snake | CU_Pguttatus_1 | GenArk |
| Tropical clawed frog | UCB_Xtro_10.0 | GenArk |
| African clawed frog | Xenopus_laevis_v10.1 | GenArk |
| Axolotl | UKY_AmexF1_1 | GenArk |
| Zebrafish | danRer11 | UCSC |
| Fugu | fr3 | UCSC |
| Nile tilapia | oreNil2 | UCSC |
| Fruitfly | Release_6_plus_ISO1_MT | GenArk |
| Yellow fever mosquito | AaegL5.0 | GenArk |
| Bombus flavifrons | iyBomFlav1 | GenArk |
| Baker’s yeast | GCA_002571405.2 | GenArk |
| Aspergillus nidulans | GCF_000149205.2 | GenArk |
| Penicillium rubens | GCF_028828025.1 | GenArk |
| Thale crees | TAIR10.1 | GenArk |
| Japanese Rice | IRGSP-1.0 | GenArk |
| Bread wheat | IWGSC_CS_RefSeq_v2.1 | GenArk |
Formatos Básicos
Ficheros de Secuencias
Los ficheros de secuencias son ficheros de texto que contienen una representación alfabética de secuencias de nucleotidos y/o proteínas. Generalmente se denominan ficheros fasta, cuando contienen una sola secuencia, o multifasta, cuando continene múltiples secuencias. Estos se pueden encontrar con diferentes extensiones como: *.fa, *.fasta o *.fas.
Cada secuencias incluída en el fichero está compuesta por una línea identificadora o cabecero, que inicia con el caracter “>” y una línea secuencia que se encuentra debajo de su identificador.

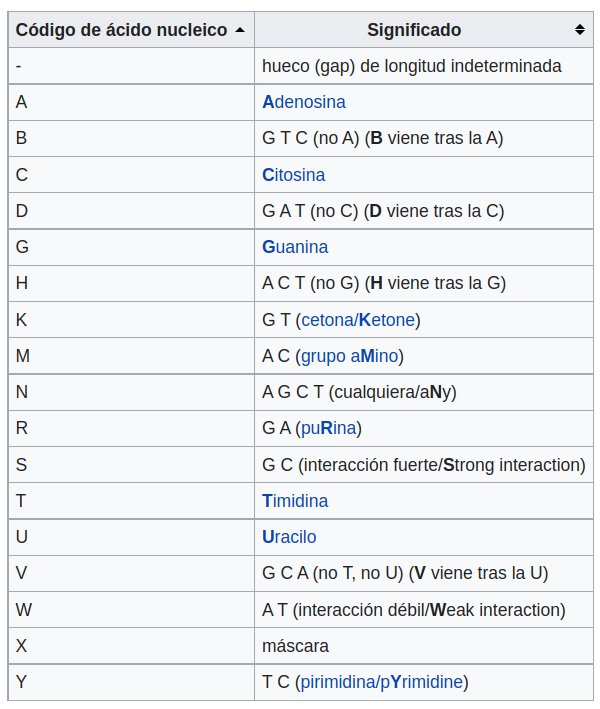
Por otro lado, cada caracter incluido en las líneas secuencias de un fichero fasta representa un nucleótido/aminoácido o caracteres ambiguos que representas múltiples opciones, como se indica en la siguiente tabla.
Anotación de regiones/elementos
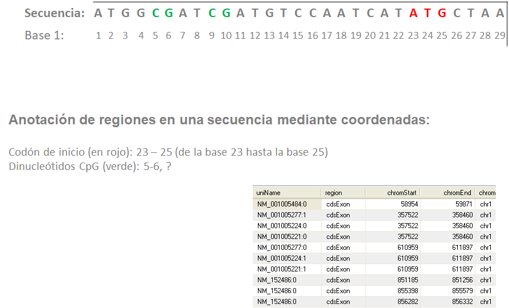
Los elementos genómicos no son más que regiones en un especio bidimensional, por lo que pueden ser identificados como dos puntos en una línea, es decir dos valores numéricos o coordenadas que representan la posición de inicio y de final del elemento en la secuencia genómica o proteínica.
A nivel de genomas las coordenadas cromosómicas se refieren a una especie, ensamblado y cromosoma determinado. Los ensamblados son versiones diferentes del genoma de una misma especie, y estas versiones se referencian con numeración ascendente, por lo tanto, cuanto más reciente sea el ensamblado mayor será la numeración de su identificador. Por ejemplo, para la especie humana (a fecha de julio 2022), el más reciente es el ensamblado GRCh38, siendo el anterior GRCh37. Además, cada uno de estos ensamblados puede presentar numerosos parches (el más reciente para el ensamblado GRCh38 es el parche 14, y se referencia a continuación del ensamblado de la siguiente manera: GRCh38.p14). La principal diferencia entre parches y ensamblados es que en los parches sólo se modifica la composición de la secuencia, mientras que entre ensamblados suele cambiar la longitud de los cromosomas, y por lo tanto de las posiciones de los elementos genómicos de los mismos. Esto se debe a que, en nuevas versiones del ensamblado de una especie, gracias a nuevos análisis de los datos crudos de secuenciación o mejoras en las técnicas de secuenciación, se consigue una mayor resolución para determinar la secuencia de regiones compactas del ADN (regiones heterocromáticas) previamente inaccesibles, o se mejora el ensamblado de elementos repetidos previamente difíciles de determinar su ubicación exacta. En la página web del NCBI podéis encontrar información detallada a este respecto: https://www.ncbi.nlm.nih.gov/grc.

Ficheros de anotación
Los ficheros de anotación de regiones genómicas más sencillos son los ficheros BED, al igual que las secuencias son ficheros de texto pero en este caso tabulados donde cada línea representa un elemento finito en una secuencia con una coordenada de inicio y otra de final.

Su extensión más habitual es *.bed, aunque pueden encontrarse con cualquier tipo de extensión. Los ficheros BED más sencillos incluyen 3 columnas con la siguiente información en orden: secuencia a la que hace referencia (cromosoma por ejemplo), coordenada de inicio y coordenada de final. Sin embargo, se pueden encontrar ficheros BED que incluyen información más compleja mediante la adición de hasta 12 columnas, que pueden utilizarse para anotar estructuras más complejas como la distribución de exones e intrones en un gen.
Las coordenadas de la anotación han heredado históricamente las especificidades de los lenguajes de programación que se usaban en el momento de su definición. En el caso de los ficheros BED, el lenguaje mayoritario cuando se definió utilizaba una anotación en base 0, por lo que las coordenadas incluídas en el fichero deben empezar a contar antes de la primera base de la secuencia (posición 0), mientras que otro tipo de anotaciones cuentan posición a posición (base 1).