A pesar de la depleción generalizada de los dinucléotidos CpG en el genoma de mamíferos, esta depleción es asimétrica, encontrando regiones densas de estos dinucleótidos conocidas como islas CpG. Las islas CpG se consideran la manifestación en el genoma de mamíferos del patrón global de metilación del ADN.

Estos elementos suelen presentar un elevado %GC y una relación de CpG observados/esperados alta, al contrario que la mayoría del genoma.

Existen 3 tipos de métodos computacionales de detección de islas CpG: métodos de ventanas deslizantes, métodos de adición y métodos de agrupamiento (clustering). Cada uno presenta sus ventajas y desventajas, y dependiendo de lo que busquemos utilizaremos unos u otros. El método clásico de detección de islas CpG es el de ventanas deslizantes (o móviles), donde se deslizará una ventana de un tamaño determinado a lo largo de la secuencia, calculando en cada paso una serie de parámetros composicionales a los que se aplicarán umbrales para determinar su pertenencia a una isla CpG.

Estos métodos de ventana presentan múltiples parámetros, pero por motivos históricos hay dos conjuntos de parámetros muy utilizados.
- Los parámetros definidos por Gardiner-Garden & Frommer (1987)
- Longitud de ventana 100 pbs
- Longitud mínima > 200 pbs
- %GC > 50%
- CpG O/E > 0.6
- Los parámetros definidos por Takai & Jones (2002)
- Longitud de ventana 200 pbs
- Longitud mínima > 500 pbs
- %GC > 55%
- CpG O/E > 0.65
Mediante el algoritmo cpgplot de EMBOSS calcularemos ambos conjuntos de islas CpG
El programa cpgplot nos devuelve tres objetos: la localización de las islas en formato cpgplot, gráficos a lo largo de la secuencia de los parámetros utilizados y la localización de las islas en formato gff2. El que nos interesa es el formato gff2, que podremos convertir a formato BED y contabilizar el número de elementos identificados.
En el menú Convert Formats usaremos la herramienta GFF-to-BED para transformar el fichero gff2 a bedEl fichero BED obtenido puede ser descargado y representado visualmente en el UCSC Genome Browser.
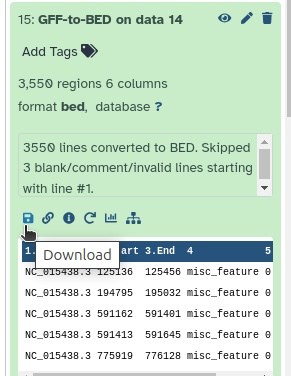
Con un editor de texto introduciremos una primera línea que comience por “track” como cabecero de nuestro fichero de anotación, para más detalles consultad los detalles del formato BED.
Accedemos al buscador de nuestro ensamblado usando la pestaña Genomes
Una vez cargado entraremos en el menú My Data > Custom Tracks
Seleccionamos nuestro fichero *.bed
Subimos el fichero presionando Submit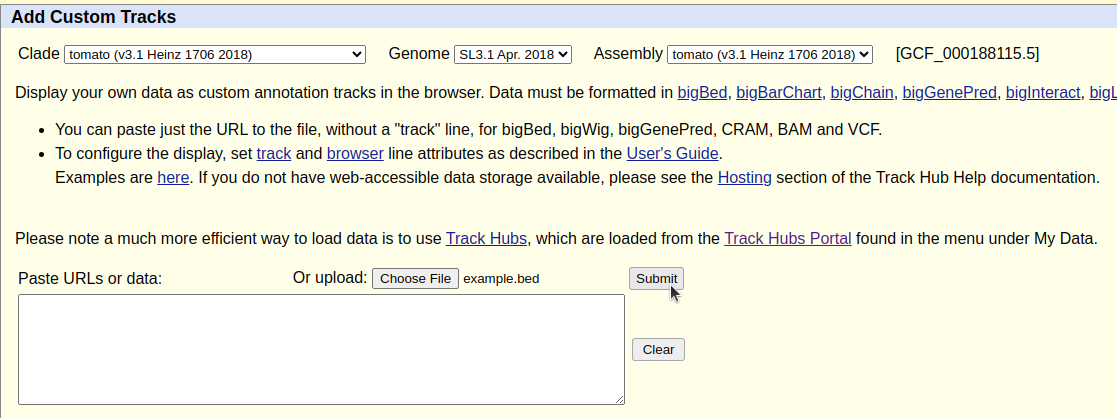
Comprobamos que nuestro fichero sea el correcto
Pinchamos en Go to first annotation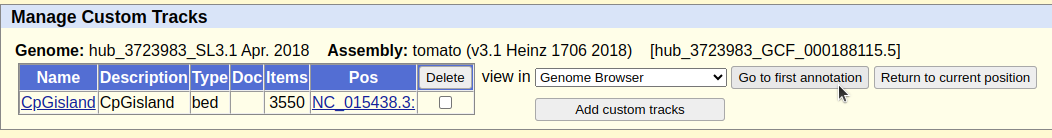
Una vez subido el fichero podemos ver nuestra nueva anotación generada en conjunto con todas las anotaciones disponibles para el ensamblado en cuestión en el buscador de la UCSC.
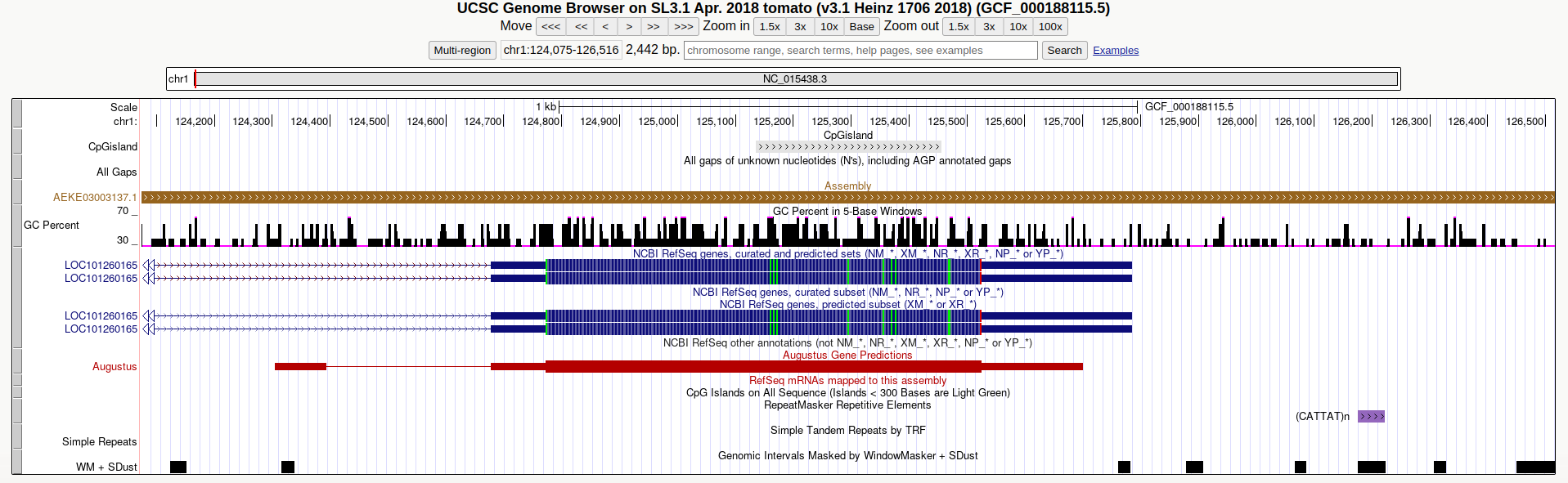
ANTES DE CONTINUAR COMPLETA EN LA TABLA COMPARTIDA:
Número de islas CpG para cada combinación de parámetros y cromosoma.