Filtrado y validación de los SNPs (SNP calling)
Para obtener información más detallada sobre cada posición del alineamiento, usaremos el programa pileup. Este programa facilita la selección e identificación de los SNPs más relevantes.
Ejecute pileup sobre las lecturas alineadas que obtuvo en el ejercicio anterior (archivo NA12878.chr22_exome.BWA_mapped.bam):
NGS: SAMtools –> Generate Pileup
Mantenga todas las opciones por defecto, excepto Call consensus according to MAQ model -> Yes. Esto genera una base de consenso en cada posición cromosómica.
Nota: MAQ es un software para el mapeado de lecturas cortas frente a ensamblados. Más información aquí.
El cambio anterior abrirá más opciones; manténgalas todas por defecto y pinche en ‘Execute‘.
Cambie a formato pileup el archivo que acaba de generar:
Herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ) –> Datatype > pileup
El archivo pileup resume los datos de las lecturas en aquellas regiones genómicas cubiertas por al menos una lectura.
Para aumentar la calidad de las variantes obtenidas, aplicaremos dos filtros sucesivos:
A. NGS: SAM Tools –> Filter Pileup
- Select ‘Pileup with ten columns (with consensus)’
- Do not report positions with coverage lower than = 10
- Convert coordinates to intervals = Yes
Con este primer filtro obtendremos unos 16.096 SNPs, lo que resulta aún muy alto. Téngase en cuenta que cabe esperar 1 SNP por kb; puesto que el exoma del cromosoma 22 tiene 600-700 kb (2% de 33500 kb, que es el tamaño de este cromosoma), cabría esperar unos 600-700 SNPs. Así que todavía son muchos.
B. Filter and Sort –> Filter
- Seleccione los SNPs que tengan ‘c7>50′. La columna 7 contiene un score que combina varias medidas de calidad, como la cobertura o la calidad por posición.
- Ahora el número de SNPs se reduce a 898, lo que no está muy lejos de lo que se espera.
Visualización en Genome Browser y validación con dbSNP
- Con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), cambie a formato pileup.
- Cambie el nombre del último archivo obtenido a: NA12878_high_confidence_SNPs. [icon name=”floppy-o” class=”” unprefixed_class=””] Save attributes.
- De nuevo con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), cambie a formato interval. [icon name=”floppy-o” class=”” unprefixed_class=””] Save attributes.
-
Display at UCSC main.
- En el navegador UCSC especifique el chr22 y active el track ‘Common SNPs’ para comparar los variantes obtenidos con los que hay anotados en la base de datos.
Ejemplos
Variante en homocigosis:
chr22:35,947,536-35,947,638
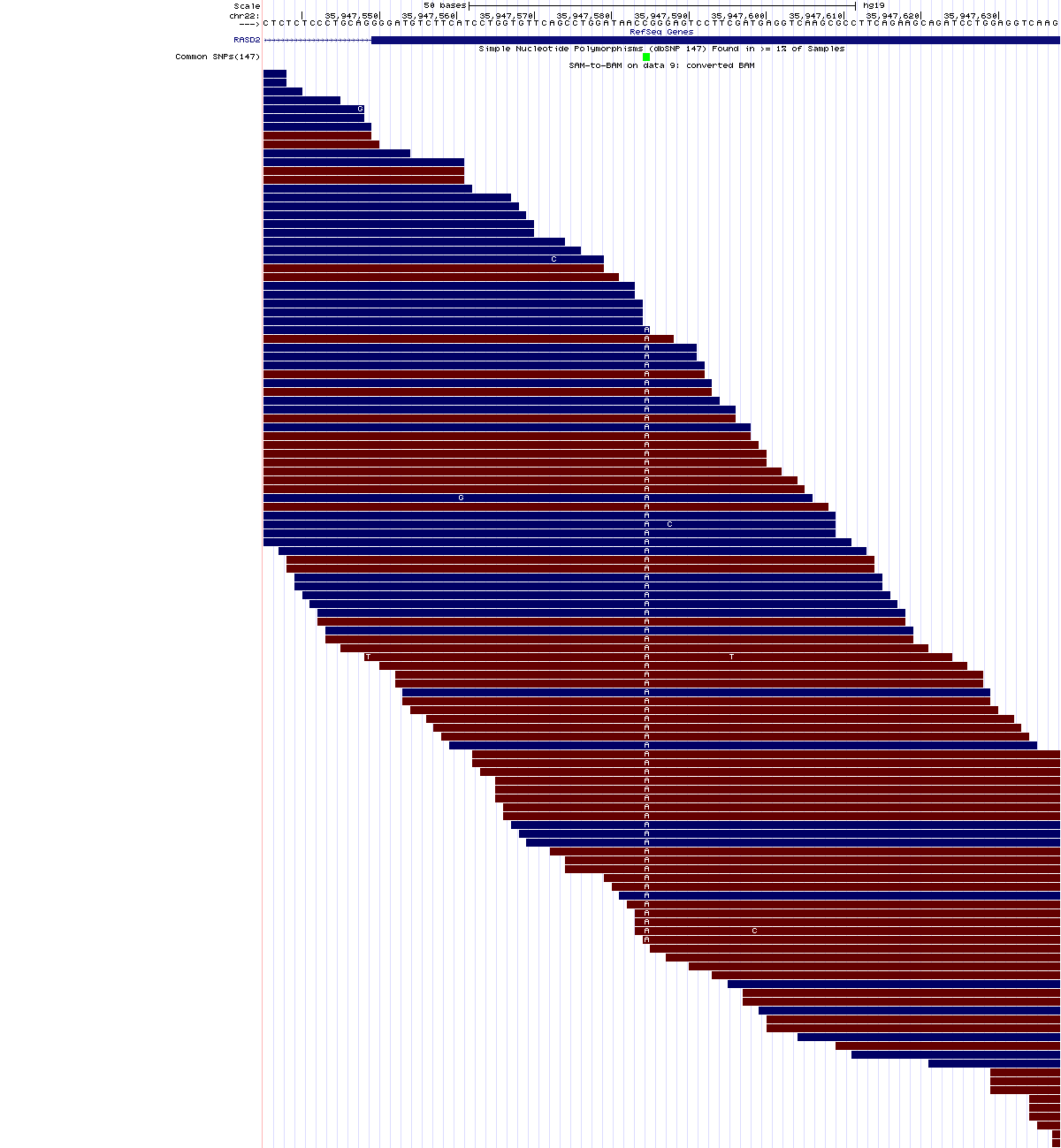
Pinchando sobre el nombre del SNP que aparece en verde en el último track (rs41279993), puede ver la anotación que hay en dbSNP sobre esta variante:

Así pues, se trata de una variante homocigótica A/A que provoca un cambio sinónimo en la primera posición de un codón que codifica para arginina.
Variante en heterocigosis
chr22:36,007,039-36,007,052
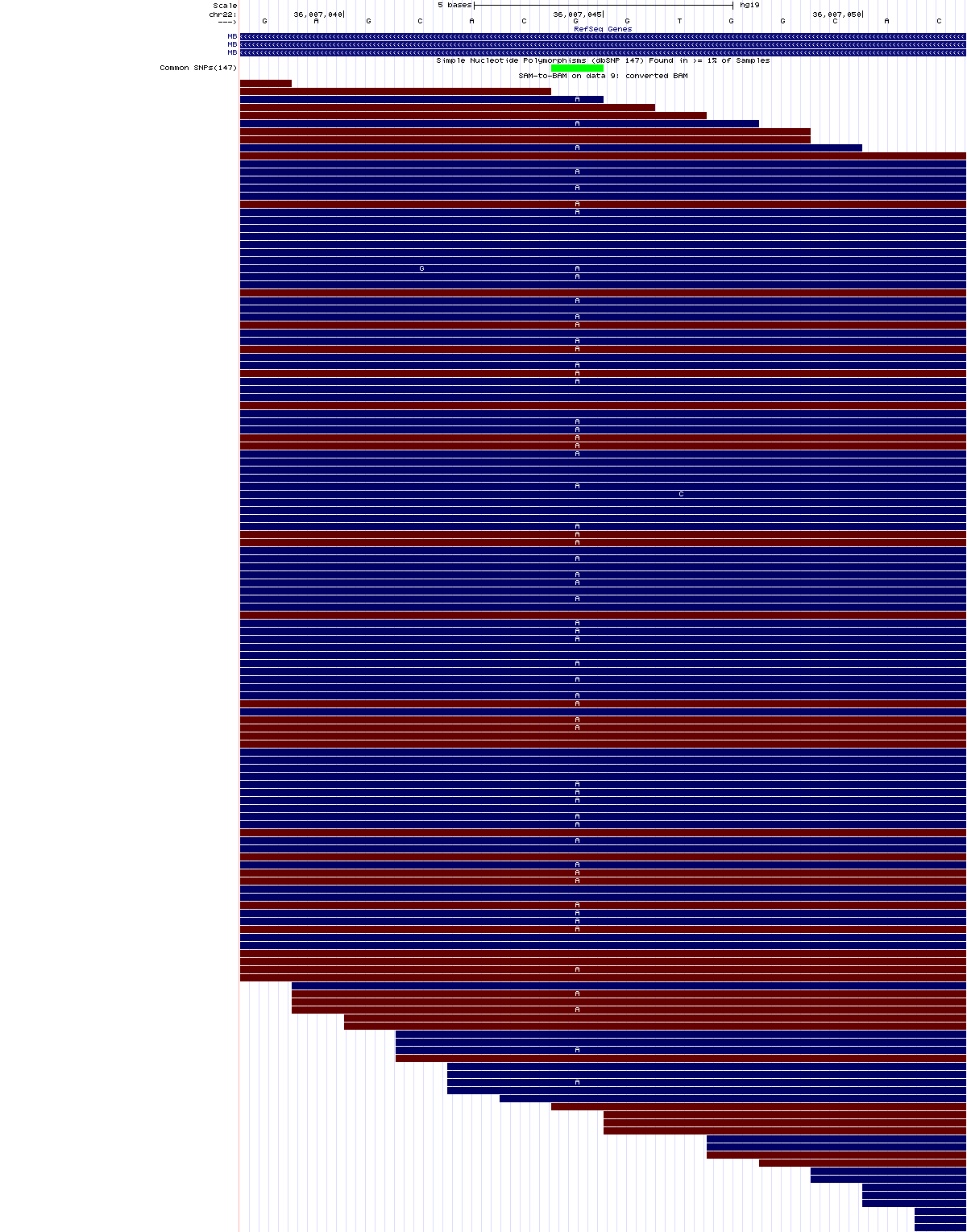
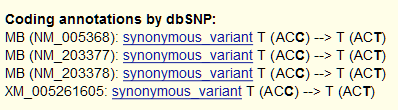
En este caso, nuestra variante (G->R) está en heterocigosis y ocurre dentro del exón 2 del gen de la mioglobina (MB) que está en la hebra complementaria inversa, y corresponde al SNP rs7292, anotado en dbSNP.