Objetivos
El objetivo de este ejercicio es la detección de variantes (SNPs e indels) en ADN genómico extraído de una única persona y enriquecido en exoma, utilizando para ello un pequeño conjunto de lecturas cortas (single-end) del cromosoma 22 obtenidas mediante NGS en un equipo Illumina GAIIx . Se trata de aproximadamente un millón de lecturas de 76bp (210 Mb en total).
El ADN procede de una mujer caucásica de los EEUU que ha sido resecuenciado multitud de veces, por lo que esta mujer es actualmente uno de los seres humanos mejor caracterizados genéticamente. Su familia fue uno de los 30 trios de los proyectos Hapmap y 1000 Genomas.
El ADN genómico se extrajo a partir de células sanguíneas, enriqueciéndolo a continuación en secuencias del exoma. Para ello, se purificaron utilizando oligonucleótidos específicos en microarrays. Este proceso de enriquecimiento no es perfecto, por lo que en la muestra final puede haber algo de contaminación de ADN no-exómico, asi como ADN mitocondrial.
Filtraremos las variantes obtenidas manualmente, de forma que se comprenda bien el procedimiento. En el mundo real, los conjuntos de datos empleados en la detección de variantes son mucho más grandes y las herramientas bastante más sofisticadas, ya que se basan en modelos estadísticos de la representación de las variantes en el conjunto de las lecturas.
Importación de los datos
- Conectese a Galaxy y entre en su cuenta (regístrese si es necesario). Servidor de Galaxy alternativo.
- Copie este enlace: http://bioinfo2.ugr.es/DatosClase/NA12878.GAIIx.exome_chr22.1E6reads.76bp.fastq.
- En el panel izquierdo de Galaxy abra Get Data -> Upload File -> Paste/Fetch data y pegue ese enlace en el cuadro de texto que aparece.
- En el campo Type elija fastqsanger, en el campo Genome elija el ensamblado Human Feb 2009 (GRCh37/ hg19) (hg19) y pulse Start.
- Cuando acaben de cargarse los datos, edítelos (herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ) en el item de la historia) y simplifique el nombre a NA12878.GAIIx.exome_chr22.1E6reads.76bp.fastq.
- Asegúrese de que el formato es fatsqsanger y el ensamblado hg19.
- Si el formato fuese otro, cámbielo editando los datos -> Datatype y elija fastqsanger.
Control de calidad
Examine las lecturas pinchando en la herramienta [icon name=”eye” class=”” unprefixed_class=””] (OJO) del item NA12878.GAIIx.exome_chr22.1E6reads.76bp.fastq.
Note que en el formato fastq cada lectura está representada por 4 líneas:
- Identificador o nombre
- Secuencia
- Separador
- Línea de calidad
Analice la calidad de las lecturas con FASTQC: En el panel izquierdo de Galaxy, seleccione NGS: QC and manipulation –> FASTQC Read Quality Reports.
Aparecerá seleccionado por defecto el fichero con los datos (extension .fastq).
Deje las demás opciones por defecto y pinche en Execute. Cuando termine el proceso, aparecerán dos items nuevos en su historia, uno con los resultados en forma numérica y otro en html (Webpage). Seleccione este último item y pinche en la herramienta [icon name=”eye” class=”” unprefixed_class=””] (OJO) para examinar los resultados.
Aparecerá un FastQC Report conteniendo una lista de gráficos con las medidas de calidad. Examine el gráfico Per base sequence quality. Observe que los datos parecen bastante buenos, la mayoría de los scores están por encima de 30, lo que corresponde a una precisión del 99.9% (véase una explicación de los Phred Quality Scores).
Note también que el gráfico Sequence Duplication Levels revela que hay una tasa de duplicación muy alta en las lecturas (artefacto debido a la PCR). Ello requeriría emplear una herramienta específica para eliminar dicha redundancia, aunque por brevedad aquí nos saltaremos ese paso.
Alineamiento
Se trata de alinear o mapear cada lectura de la muestra de ADN (NA12878.GAIIx.exome_chr22.1E6reads.76bp.fastq) con el genoma de referencia (hg19), de forma que podamos identificar las variantes (SNVs, SNPs o indels).
NGS: Mapping –> Map with BWA for Illumina:
- Use el fichero FASTQ por defecto (NA12878.GAIIx.exome_chr22.1E6reads.76bp.fastq)
- Seleccione Human (Homo sapiens) (b37): hg19 como genoma de referencia
- Deje las demás opciones por defecto
- Pinche en Execute
Este es el paso que más tarda (5-20 minutos, dependiendo de la carga que tenga el servidor de Galaxy en ese momento).
Cuando termine el proceso, pinche en la herramienta [icon name=”eye” class=”” unprefixed_class=””] (OJO) y examine el alineamiento en formato SAM (Sequence Alignment Map).
Observe que muchas lecturas no mapean en el cromosoma 22 (columna 3). Para quedarnos solamente con aquellas lecturas que mapean correctamente en el cromosoma, debemos filtrar estos resultados:
- Filter and Sort –> Filter
- Condition: c3==’chr22′
En el nuevo item de su historia, solo aparecerán ahora aquellas lecturas que mapean en el cromosoma 22 (un 93% de las lecturas).
Con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), renombre este último item a: NA12878.chr22_exome.BWA_mapped.chr22_filtered
Ahora debemos comprimir el archivo en formato SAM, poniéndolo en formato binario (BAM):
NGS: Sam Tools –> SAM-to-BAM
Asegúrese de que selecciona para ello el item más reciente en su historia (NA12878.chr22_exome.BWA_mapped.chr22_filtered) y pinche en ‘Execute‘.
Con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), renombre el archivo BAM recién obtenido a: NA12878.chr22_exome.BWA_mapped.bam
Visualización
Para visualizar el alineamiento en el UCSC Genome Browser, pinche en el nombre del archivo BAM y elija display at UCSC main.
Si activa el track de genes RefSeq y pone las coordenadas chr22:35,790,491-35,827,565 en el cuadro de texto del Genome Browser obtendrá una imagen como esta:
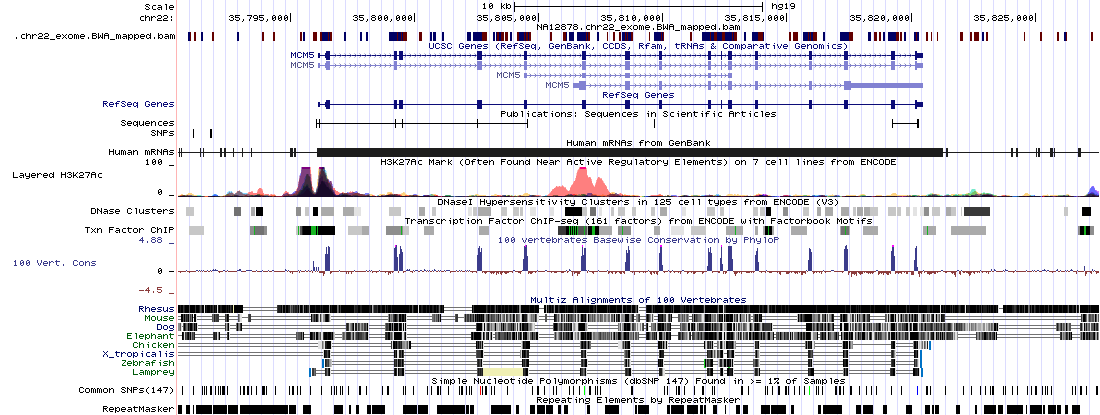
Observe como, al tratarse de datos del exoma, las lecturas se concentran en los exones.
Aumentando el zoom puede observar las variantes con respecto al genoma de referencia.
Filtrado y validación de los SNPs (SNP calling)
Para obtener información más detallada sobre cada posición del alineamiento, usaremos el programa pileup. Este programa facilita la selección e identificación de los SNPs más relevantes.
Ejecute pileup sobre las lecturas alineadas que obtuvo en el ejercicio anterior (archivo NA12878.chr22_exome.BWA_mapped.bam):
NGS: SAMtools –> Generate Pileup
Mantenga todas las opciones por defecto, excepto Call consensus according to MAQ model -> Yes. Esto genera una base de consenso en cada posición cromosómica.
Nota: MAQ es un software para el mapeado de lecturas cortas frente a ensamblados. Más información aquí.
El cambio anterior abrirá más opciones; manténgalas todas por defecto y pinche en ‘Execute‘.
Cambie a formato pileup el archivo que acaba de generar:
Herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ) –> Datatype > pileup
El archivo pileup resume los datos de las lecturas en aquellas regiones genómicas cubiertas por al menos una lectura.
Para aumentar la calidad de las variantes obtenidas, aplicaremos dos filtros sucesivos:
A. NGS: SAM Tools –> Filter Pileup
- Select ‘Pileup with ten columns (with consensus)’
- Do not report positions with coverage lower than = 10
- Convert coordinates to intervals = Yes
Con este primer filtro obtendremos unos 16.096 SNPs, lo que resulta aún muy alto. Téngase en cuenta que cabe esperar 1 SNP por kb; puesto que el exoma del cromosoma 22 tiene 600-700 kb (2% de 33500 kb, que es el tamaño de este cromosoma), cabría esperar unos 600-700 SNPs. Así que todavía son muchos.
B. Filter and Sort –> Filter
- Seleccione los SNPs que tengan ‘c7>50′. La columna 7 contiene un score que combina varias medidas de calidad, como la cobertura o la calidad por posición.
- Ahora el número de SNPs se reduce a 898, lo que no está muy lejos de lo que se espera.
Visualización en Genome Browser y validación con dbSNP
- Con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), cambie a formato pileup.
- Cambie el nombre del último archivo obtenido a: NA12878_high_confidence_SNPs. [icon name=”floppy-o” class=”” unprefixed_class=””] Save attributes.
- De nuevo con la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ), cambie a formato interval. [icon name=”floppy-o” class=”” unprefixed_class=””] Save attributes.
-
Display at UCSC main.
- En el navegador UCSC especifique el chr22 y active el track ‘Common SNPs’ para comparar los variantes obtenidos con los que hay anotados en la base de datos.
Ejemplos
Variante en homocigosis:
chr22:35,947,536-35,947,638
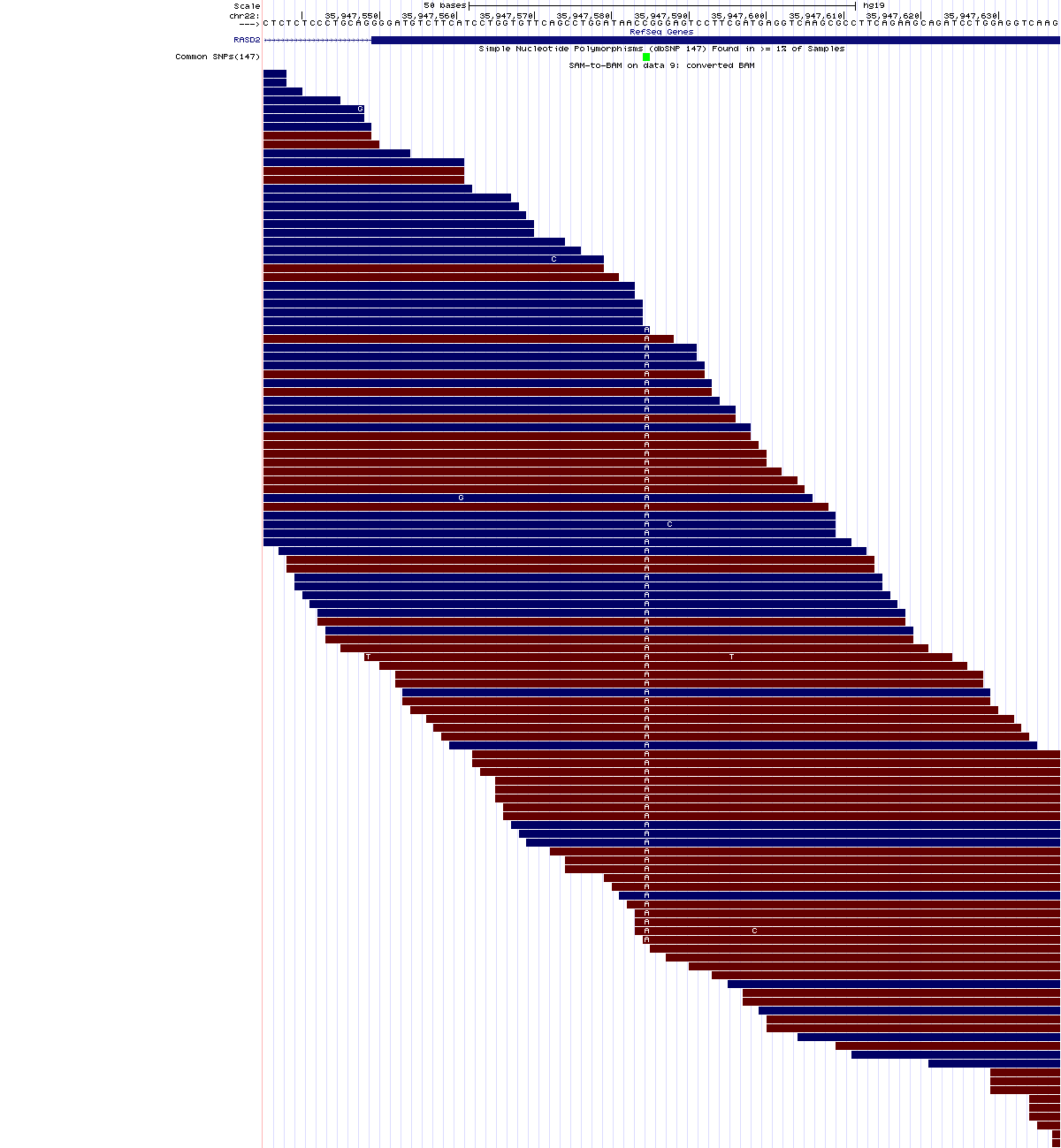
Pinchando sobre el nombre del SNP que aparece en verde en el último track (rs41279993), puede ver la anotación que hay en dbSNP sobre esta variante:

Así pues, se trata de una variante homocigótica A/A que provoca un cambio sinónimo en la primera posición de un codón que codifica para arginina.
Variante en heterocigosis
chr22:36,007,039-36,007,052
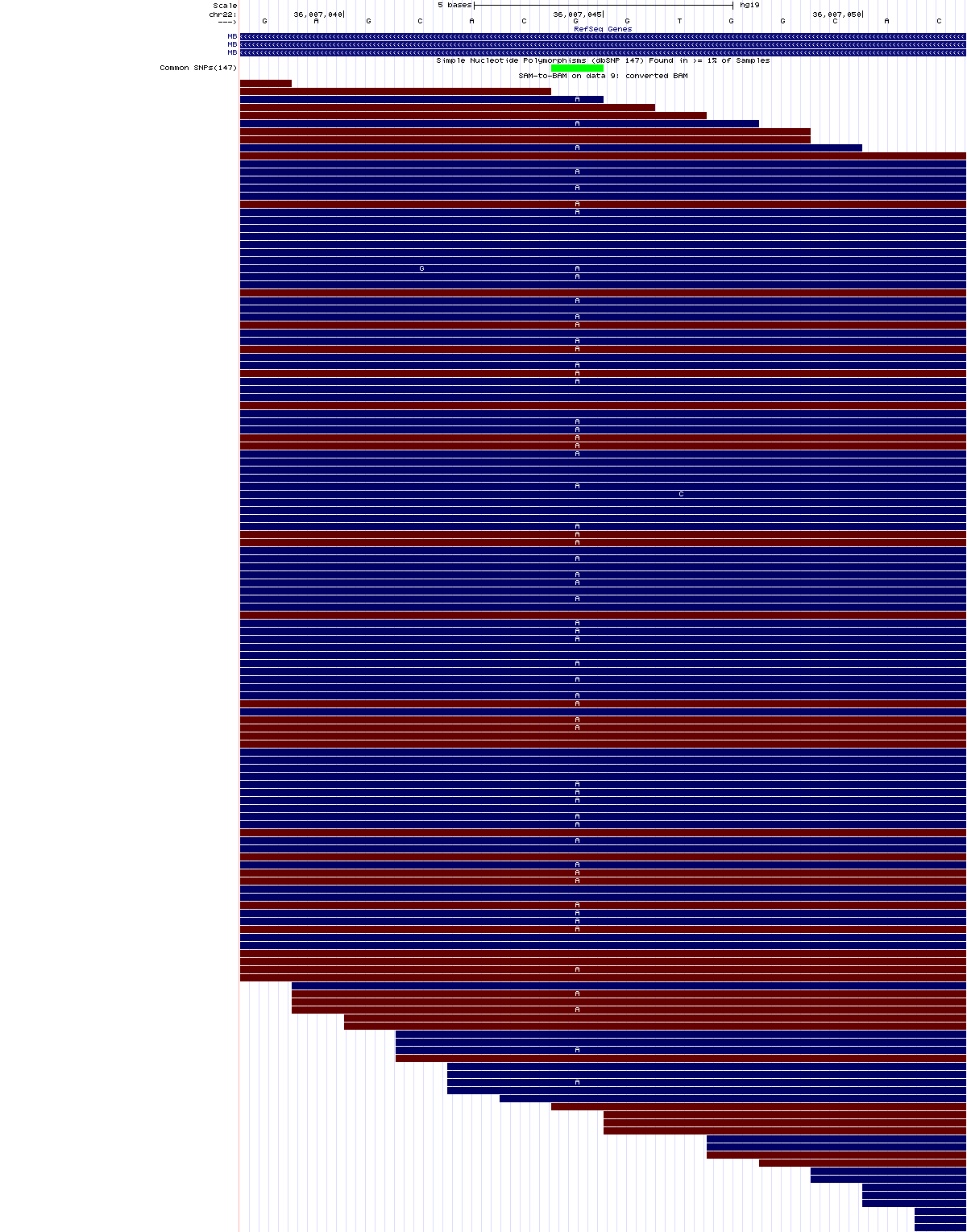
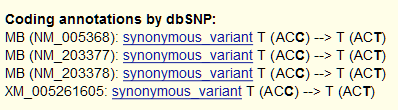
En este caso, nuestra variante (G->R) está en heterocigosis y ocurre dentro del exón 2 del gen de la mioglobina (MB) que está en la hebra complementaria inversa, y corresponde al SNP rs7292, anotado en dbSNP.
Efectos biológicos de las variantes
Para estudiar los efectos biológicos de las 898 variantes obtenidas en el ejercicio anterior, utilizaremos SnpEff. Este software permite anotar las variantes y predecir sus efectos biológicos.
En primer lugar, es necesario poner el listado de variantes en formato vcf. Pinche en la herramienta [icon name=”pencil” class=”” unprefixed_class=””] (LÁPIZ) del item NA12878_high_confidence_SNPs -> Datatype -> vcf.
Abra NGS Variant Analysis -> SnpEff Variant effect and annotation y seleccione NA12878_high_confidence_SNPs. Deje todas las opciones por defecto y pulse en Execute.
Esto generará dos nuevos items en su historia. Pinche en la herramienta [icon name=”eye” class=”” unprefixed_class=””] (OJO) del último item (SnpEff on data – stats) para examinar los diferentes resultados que este software genera, entre ellos la distribución de los efectos en las distintas regiones genómicas:
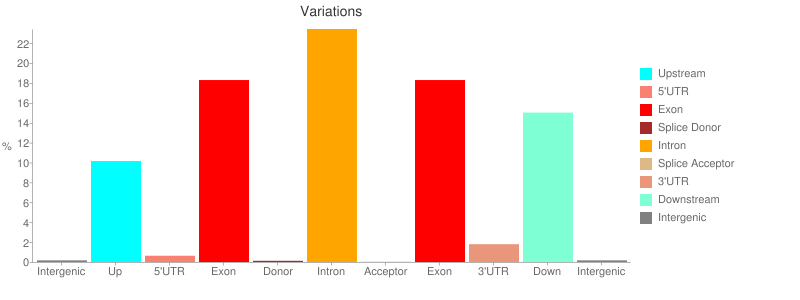
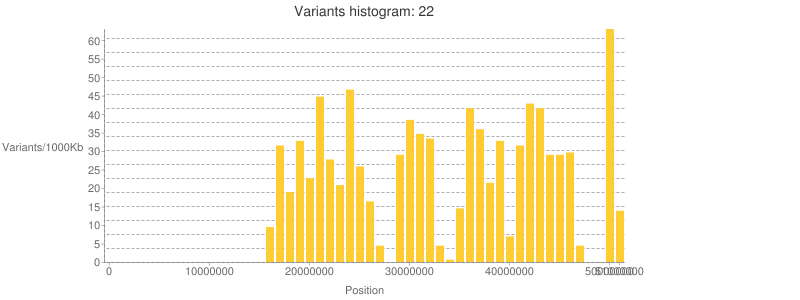
O su ditribución espacial a lo largo del cromosoma: