Los factores de transcripción son los principales responsables de la regulación génica en eucariotas, y la expansión de su repertorio se ha asociado con el desarrollo de los linajes multicelulares y el aumento de la complejidad celular. La evolución de la regulación mediada por factores de transcripción depende de múltiples elementos, como la aparición de dominios de unión al ADN, la regulación de su propia expresión o la afinidad por sus sitios de unión. En este caso, asumimos que la importancia de un factor de transcripción en una especie se correlaciona con el número de sus sitios de unión, posiblemente debido a un proceso de expansión y selección positiva de estos sitios, aunque esta suposición no necesariamente sea cierta. Por lo tanto, analizaremos la fracción del genoma cubierta por sitios de unión a factores de transcripción. Para algunas especies, existen anotaciones más o menos detalladas de sitios de unión a factores de transcripción verificados experimentalmente, como en el caso de ORegAnno, o datos experimentales de unión de proteínas al ADN (como CLIP-Seq o ChIP-Seq). Sin embargo, no todas las especies que estudiamos cuentan con este tipo de información. Por lo tanto, en estos casos, cuantificaremos el número y la densidad de los TFBS basándonos en predicciones derivadas de motivos de unión de diferentes factores de transcripción.
Sitios de Unión a Factores de Transcripción (TFBS)
Un motivo de unión de un TF al ADN es una secuencia probabilística compuesta por una matriz de frecuencias donde cadaUn motivo de unión de un factor de transcripción (TF) al ADN es una secuencia probabilística representada por una matriz de frecuencias, donde cada posición del motivo contiene la frecuencia de cada nucleótido observada en las secuencias con las que el TF ha interactuado. Esta matriz refleja la probabilidad de que un nucleótido específico se encuentre en cada posición del motivo, basada en todas las secuencias experimentales en las que se ha detectado la unión entre el TF y el ADN.
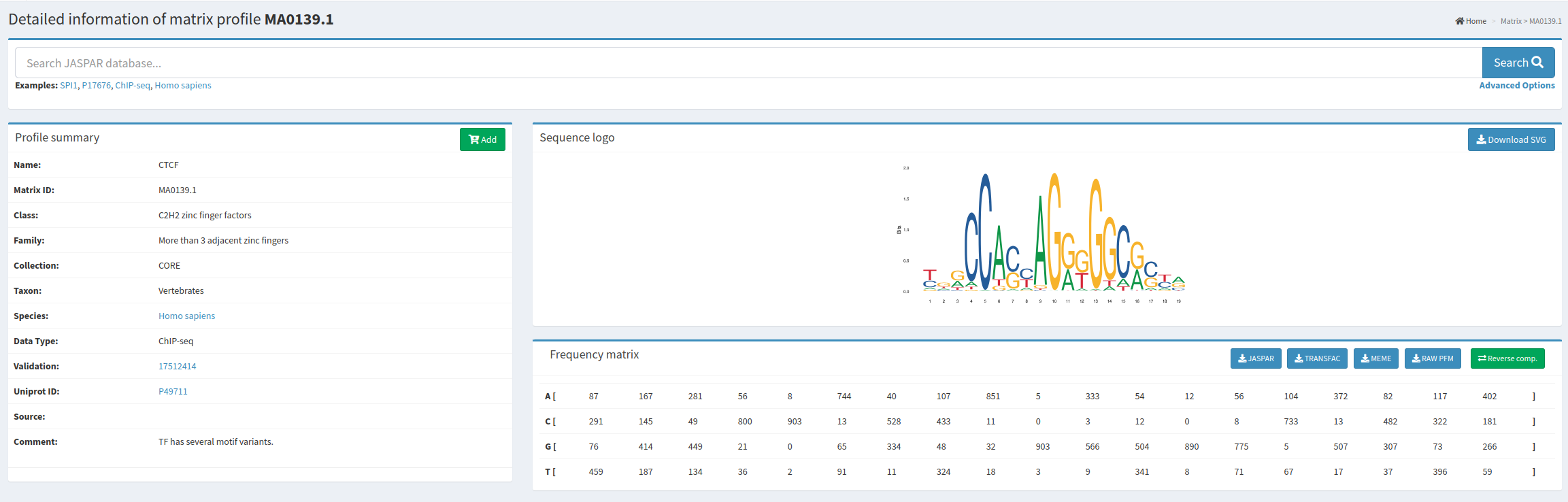
Utilizaremos la base de datos JASPAR, en la que los motivos de unión a los diferentes factores de transcripción se clasifican en seis grupos taxonómicos: vertebrados, urocordados, insectos, nematodos, hongos y plantas. Esto se debe a que, aunque el repertorio de factores de transcripción, su afinidad y sus dominios de unión al ADN suelen estar altamente conservados entre organismos relacionados, diferentes grupos taxonómicos pueden presentar repertorios de factores de transcripción muy distintos. Incluso, los mismos factores de transcripción pueden mostrar diferentes dominios de unión al ADN en grupos taxonómicos distintos.
Predicción de TFBS
Por lo general, dependiendo de la especie en estudio, utilizaríamos los modelos probabilísticos estimados específicamente para esa especie. Sin embargo, a medida que nos alejamos de los humanos en términos evolutivos, la calidad de la anotación y el número de experimentos disminuyen. Por esta razón, en estas prácticas, y asumiendo la conservación de los sitios de unión a factores de transcripción, utilizaremos los motivos de unión de humanos para el resto de las especies del estudio.
En esta práctica, nos enfocaremos en estudiar la distribución de los sitios de unión para los factores de transcripción CTCF y p53. Ambos factores están altamente conservados en los eucariotas y desempeñan funciones esenciales para el correcto funcionamiento celular. CTCF juega un papel clave en la reorganización de la cromatina y la regulación de la impronta génica, mientras que p53, conocido como el “guardián del genoma”, es crucial para prevenir la propagación de células con daño en el ADN. Las funciones de CTCF en la arquitectura del genoma y de p53 en la regulación del ciclo celular son vitales en eucariotas.
En la base de datos JASPAR, buscaremos los motivos de unión en Homo sapiens para los dos factores de transcripción propuestos.

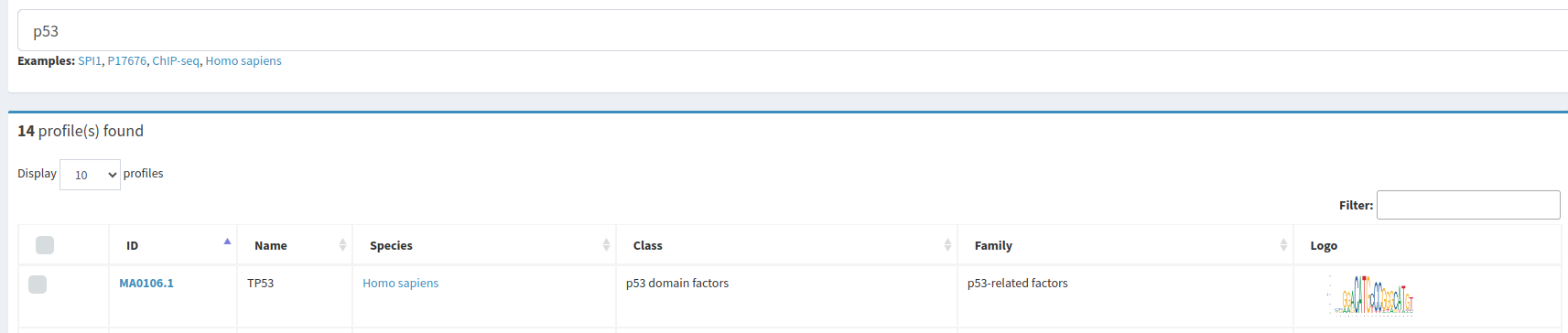
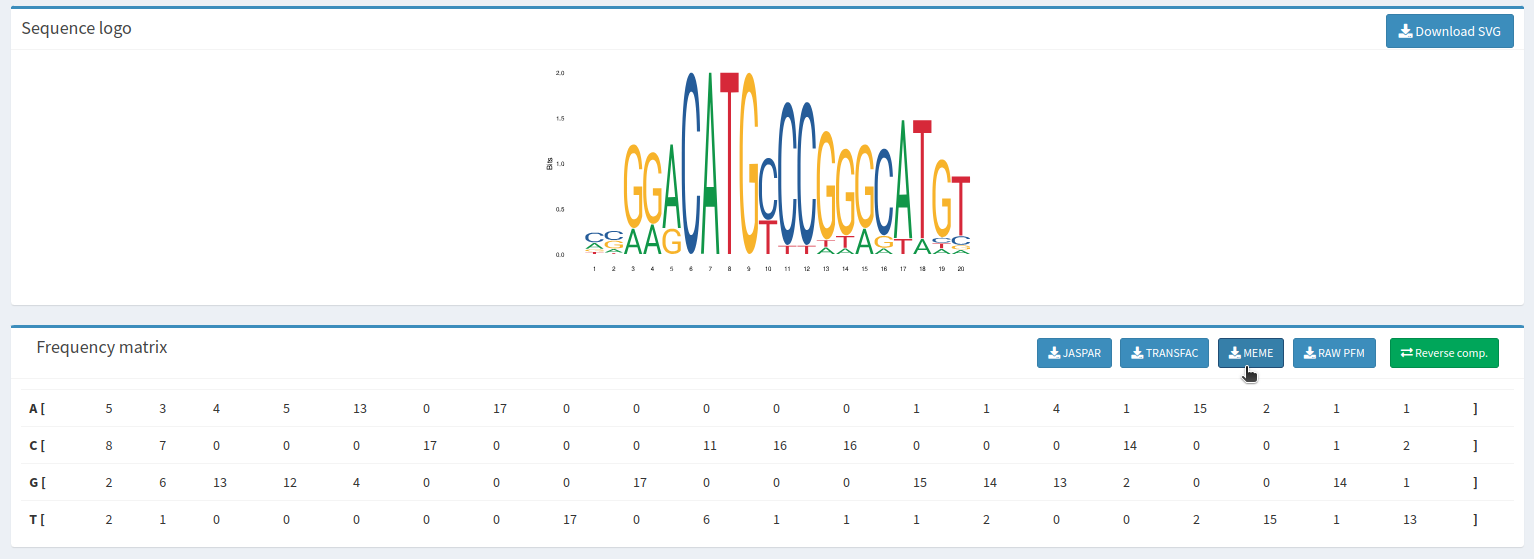
Y descargaremos el fichero meme correspondiente a cada uno de los TFBS.
Get Data > Upload File y subimos los ficheros descargados en formato memexml
Usaremos la herramienta FIMO del menú Motif para identificar los sitios de unión, los parámetros a modificar serán:
- Check reverse complement strand = Yes
- Output threshold for displaying search results = 0.00001
- Report best match in case of overlapping matches on both strands? = Yes
- Maximum number of scores that will be stored = 1000000
- Disable q-value calculation? = Yes
- I certify that I am not using this tool for commercial purposes = Yes
Modificamos el objeto resultante con la herramienta Cut del menú Text manipulation, reorganizando las columnas en el siguiente orden: c3,c4,c5,c2,c7,c5
Y una vez obtenemos el resultado podemos calcular la longitud de los TFBS con la herramienta Summary Statistics del menú Statistics
ANTES DE CONTINUAR:
Calcula y completa en las tabla compartida los valores de densidad para los sitios de unión de ambos factores de transcripción.
Expansión de TFBS mediada por elementos repetidos
Los elementos repetidos, en particular los elementos transponibles, son una fuente importante de nuevos mecanismos de regulación génica en eucariotas. Estos organismos han desarrollado numerosos mecanismos para mantener silenciados a dichos elementos. Sin embargo, muchos de ellos se transcriben y pueden modificar la expresión génica tanto en cis como en trans. Por otro lado, se ha observado que estos elementos transponibles contienen sitios de unión a factores de transcripción funcionales que se integran en las redes de regulación génica de los organismos. Interesantemente, la expansión de los sitios de unión para un factor de transcripción específico en un organismo suele proceder de una o pocas familias de elementos transponibles que contenían el TFBS en su secuencia ancestral. Por lo tanto, su expansión a lo largo del genoma aumenta el número de sitios de interacción para ese factor de transcripción. Generalmente, los elementos LTR contribuyen más que otros elementos transponibles a esta expansión de TFBS, ya que tienden a conservar su región reguladora intacta.
En el caso de los factores de transcripción p53 y CTCF, se ha demostrado que la expansión de sus sitios de unión ha sido mediada en algunas especies por la expansión de elementos Alus (SINEs) y retrotransposones de ADN, respectivamente.
Para estudiar su posible expansión a través de elementos repetidos calcularemos la densidad de estos TFBS dentro de los elementos repetidos anotados en el apartado anterior.
Mediante la herramienta intersect del menú Operate on Genomic Intervals
- Seleccionamos el objeto con los TFBS y el que contiene los elementos repetidos
- Seleccionameos Overlapping pieces of intervals
- for at least = 1
Posteriormente calcularemos la longitud total de TFBS solapando elementos repetidos mediante la herramienta Summary Statistics
Dividiendo dicha longitud por la longitud total de los TFBS obtendremos la fracción de TFBS contenida en elementos repetidos.
Completad la tabla compartida con la fracción de cada TFBS en elementos repetidos