Jupyter Notebook es una aplicación que permite crear y compartir documentos que contengan códigos, ecuaciones, visualizaciones y textos narrativos a tiempo real. Es un entorno de edición de texto y programación interactivo, que permite ejecutar líneas de código e instantáneamente obtener su resultado. Permite trabajar con numerosos lenguajes de programación como son Julia, Python o R. Sus usos son múltiples: limpieza y transformación de datos, simulación numérica, modelado estadístico, visualización de datos, machine learning y mucho más.
Recientemente, Grüning y colaboradores han logrado integrar una herramienta tan útil como es Jupyter Notebook en un entorno bastante familiar para los bioinformáticos como es Galaxy. Esta integración combina los análisis estándares de datos de Galaxy, con la capacidad de explorar de forma interactiva estos datos que nos proporciona Jupyter Notebook. Esto otorga al usuario la capacidad de obtener figuras destinadas a formar parte de una publicación a partir de datos complejos.
Práctica guiada: Jupyter + Galaxy. Emulación del programa Compseq de EMBOSS
Con el uso de la integración de Jupyter en Galaxy, vamos a reproducir el programa Compseq de EMBOSS para una secuencia de DNA determinada. Es decir, vamos a calcular la composición en dinucleótidos de una secuencia de DNA, a comparar esta composición con la esperada estadísticamente y a obtener figuras que representen nuestros resultados.
En primer lugar, debemos acceder a la página de Galaxy (https://usegalaxy.org/), realizar un log in con nuestro usuario y contraseña y crear una nueva historia. Seguidamente, usamos la herramienta USCS main table browser, en el grupo Get data, para obtener la secuencia de DNA problema. En esta práctica usaremos la secuencia del gen FAM81A, que sitúa su entorno genómico entre las posiciones 59723573 y 59821999 del cromosoma 15 (chr15:59723573-59821999). Es importante que el fichero que obtengamos tenga formato FASTA simple, para ello seleccionamos como formato de salida sequence.
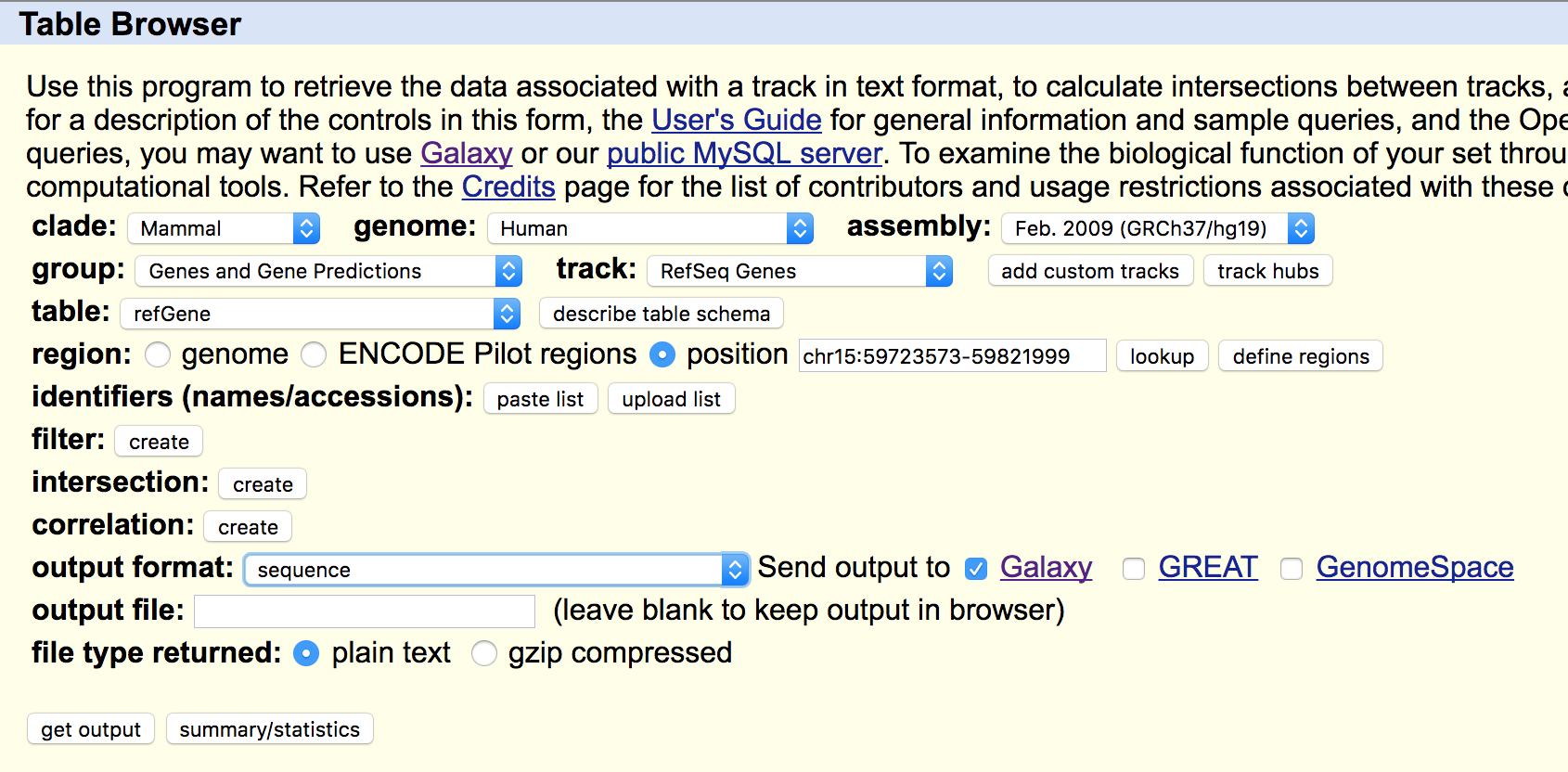
Esto nos llevará a un nuevo menú el que tenemos que indicar el tipo de secuencia para los genes RefSeq, seleccionamos ‘genomic’ en nuestro caso.

Por último se nos abre un menú que nos permite seleccionar la región genómica que queremos obtener, esto es arbitrario, y el formato de secuencia. Seleccionamos ‘All upper case’, para tener nuestra secuencia genómica en mayúsculas.

Una vez cargada la secuencia en Galaxy debemos abrir el entorno de programación Jupyter Notebook. Para ello abrimos la pestaña Visualizations del Menú superior de Galaxy y seleccionamos la opción ‘Interactive Environment’.
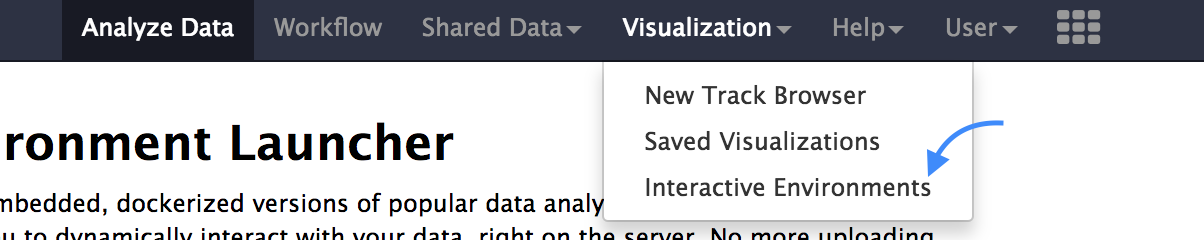
Seleccionamos Jupyter como entorno, usamos su imagen por defecto y cargamos nuestro set de datos.

Y así obtendremos una pantalla como esta:
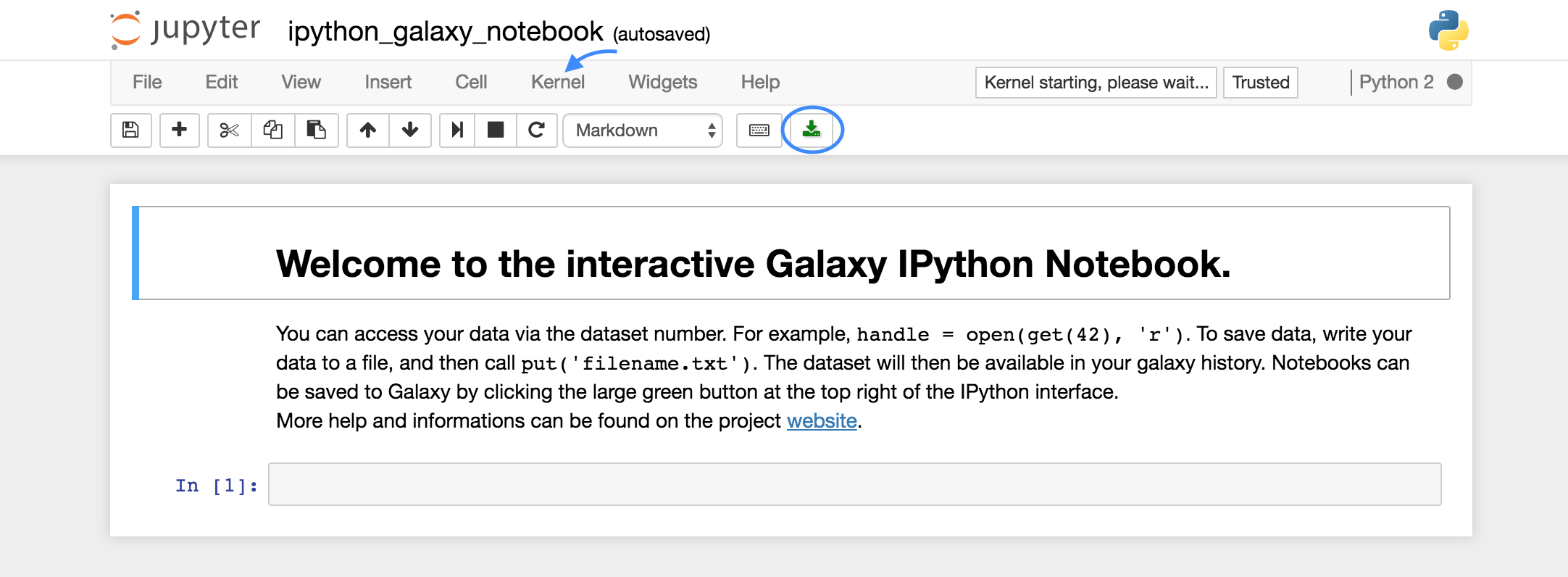
Esta es la interfaz normal de Jupyter Notebook, lo primero que vamos a realizar es seleccionar Python 3 como el lenguaje de programación activo. Para ello abrimos la pestaña ‘Kernel’, seleccionamos ‘Change Kernel’ y escogemos Python 3.
El botón verde señalado en la imagen es el que nos permite exportar nuestro Notebook a Galaxy para que aparezca en nuestra historia.
Como podemos observar, en Jupyter Notebook se trabaja con celdas. Estas celdas tienen asignado un tipo que podemos cambiar cuando queramos. Por ejemplo, las celdas destinadas a contener texto son del tipo markdown y las que están destinadas a tener código son del tipo code. Las celdas de tipo code poseen a su izquierda una anotación del tipo In [n]. Esta anotación indica el orden en que se están ejecutando las diferentes celdas, pues no tiene porqué ser de arriba a abajo.
Para ejecutar el contenido de una celda code se puede abrir la pestaña Cells y seleccionar alguna de las opciones que contienen Run Cell, o bien usar el atajo mayus + intro.
Una vez entendido lo básico sobre Jupyter Notebook, vamos a reproducir el programa Compseq.
Para ello debemos cargar nuestro archivo de Galaxy en Jupyter. Lo realizamos con el siguiente comando:
f = open(get(n))
Donde n es el número que posee el archivo en nuestra historia, debería ser 1 en nuestro caso. Básicamente vamos a usar una ventana móvil de tamaño 2 que va a ir contando los dinucleótidos de nuestra secuencia. Para ello existen dos factores que nos molestan en el formato FASTA original: la línea de cabecera, y la división de la secuencia en líneas. Para transformar nuestro fichero a un formato adecuado para el uso de la ventana móvil usamos las siguientes líneas de comando:
seq = ""
for linea in f:
if not linea.startswith('>'):
seq += linea.strip()
print(seq)
Este bucle añade todas las líneas que no empiezan por ‘>’ a la variable seq. Además, la función strip() elimina los saltos de línea, por lo que obtenemos la secuencia de nucleótidos en una sola línea.
A continuación debemos contar los distintos dinucleótidos. Para ello vamos a definir una variable tipo diccionario. La variable tipo diccionario, al igual que este último contiene palabra – significado, contiene datos tipo clave – valor. En nuestro caso cada clave será un dinucleótido (ej: ‘AA’ o ‘CG’) y su valor asignado deberá ser la cantidad de ese tipo de dinucleótido encontrada en nuestra secuencia. Usamos el siguiente código:
obsrv = dict()
for n in range(len(seq)-1):
dinuc = seq[n:n+2]
if not dinuc in obsrv:
obsrv[dinuc] = 1
else:
obsrv[dinuc] += 1
print(obsrv)
Este bucle recorre todas las posiciones de la secuencia menos la última (que no tendría dinucleótido asociado). La variable dinuc toma como valor la letra de esa posición en la secuencia y y la letra de la posición contigua. Tomando así como valor ‘GC’ por ejemplo. Si ‘GC’ no está como clave nuestro diccionario, entonces la toma como clave y le otorga el valor 1, si sí estaba, incrementa su valor en 1. De esta forma, cada vez que encontremos ‘GC’ sumaremos 1 a su valor en nuestro diccionario.
Así obtenemos un diccionario que contiene el número total de cada dinucleótido en nuestra secuencia. Sin embargo, nuestro diccionario está desordenado, tendrá el orden en que se han ido introduciendo las claves. Para solucionar esto usamos una función de ordenado del paquete collections de Python. El comando sería el siguiente:
import collections obsrv = collections.OrderedDict(sorted(obsrv.items()))
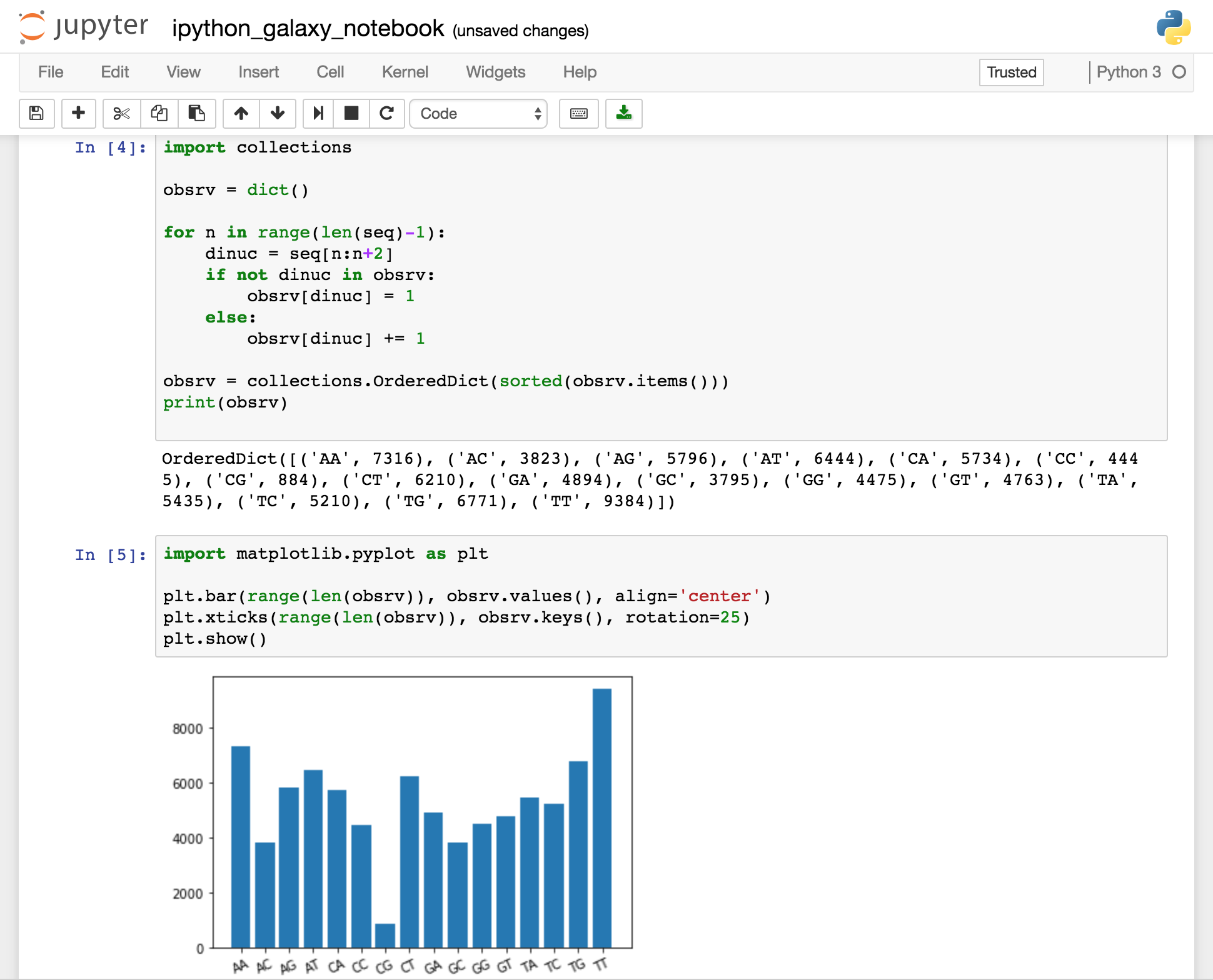
Una vez obtenidos los datos de los dinucleótidos contenidos en la secuencia, procederemos a representarlos en un histograma. Para ello usaremos el paquete de Python matplotlib.pyplot. El código es el siguiente:
import matplotlib.pyplot as plt plt.bar(range(len(obsrv)), obsrv.values(), align='center') plt.xticks(range(len(obsrv)), obsrv.keys(), rotation=25) plt.show()
Por partes, plt.bar va a dibujar las distintas barras del histograma. Para ello requiere un rango de números (para el eje x) en los que va a ir cada barra. En nuestro caso este rango va de 1 a 16, uno por cada dinucleótido. La función range genera un rango de n números, n en este caso es el tamaño de nuestro diccionario (16 claves). En segundo lugar requiere los valores del eje y de cada barra, que no son otros que los valores asignados a nuestras claves en el diccionario. plt.xticks nos permite establecer las etiquetas del eje x del mismo modo que situamos las barras. Por último mostramos la figura con plt.show().
Así si todo ha ido bien deberíamos obtener lo siguiente:
A continuación calcularemos los dinucleótidos esperados estadísticamente. La frecuencia de aparición de un dinucleótido puede calcularse como el producto de las frecuencias de los nucleótidos que lo forman. Así la frecuencia del dinucleótidos ‘CG’ por ejemplo, será igual al producto entre la frecuencia de ‘C’ y la frecuencia de ‘G’ en nuestra secuencia. Este valor obtenido deberá multiplicarse por el total de dinucleótidos en la secuencia, que será el número total de nucleótidos menos uno, el último.
¿Cómo hallamos las frecuencias de nucleótidos? Pues simplemente dividiendo el total de cada tipo de nucleótido de nuestra sequencia, por el número de nucleótidos obtenidos en la misma.
El código para calcular los dinucleótidos esperados y guardarlos en un diccionario como hicimos con los observados, es el siguiente:
import collections
count = dict()
total_nuc=0
for n in range(len(seq)):
nuc = seq[n]
total_nuc += 1
if not nuc in count:
count[nuc] = 1
else:
count[nuc] += 1
esper = dict()
for key in count:
for key2 in count:
esper[key+key2] = count[key] / total_nuc * count[key2] / total_nuc * (total_nuc - 1)
esper = collections.OrderedDict(sorted(esper.items()))
print(esper)
Como podemos ver, nos servimos de un diccionario intermedio (count) para obtener el número de cada tipo de nucleótido contenido en la secuencia. Para obtener el diccionario esper, que contiene los dinucleótidos esperados, realizamos un doble bucle que emparejará cada tipo de nucleótido con los cuatro tipos de nucleótidos para realizar la operación anteriormente descrita.
Seguidamente realizamos un plot exactamente igual que para los observados:
import matplotlib.pyplot as plt plt.bar(range(len(esper)), esper.values(), align='center') plt.xticks(range(len(esper)), esper.keys(), rotation=25) plt.show()
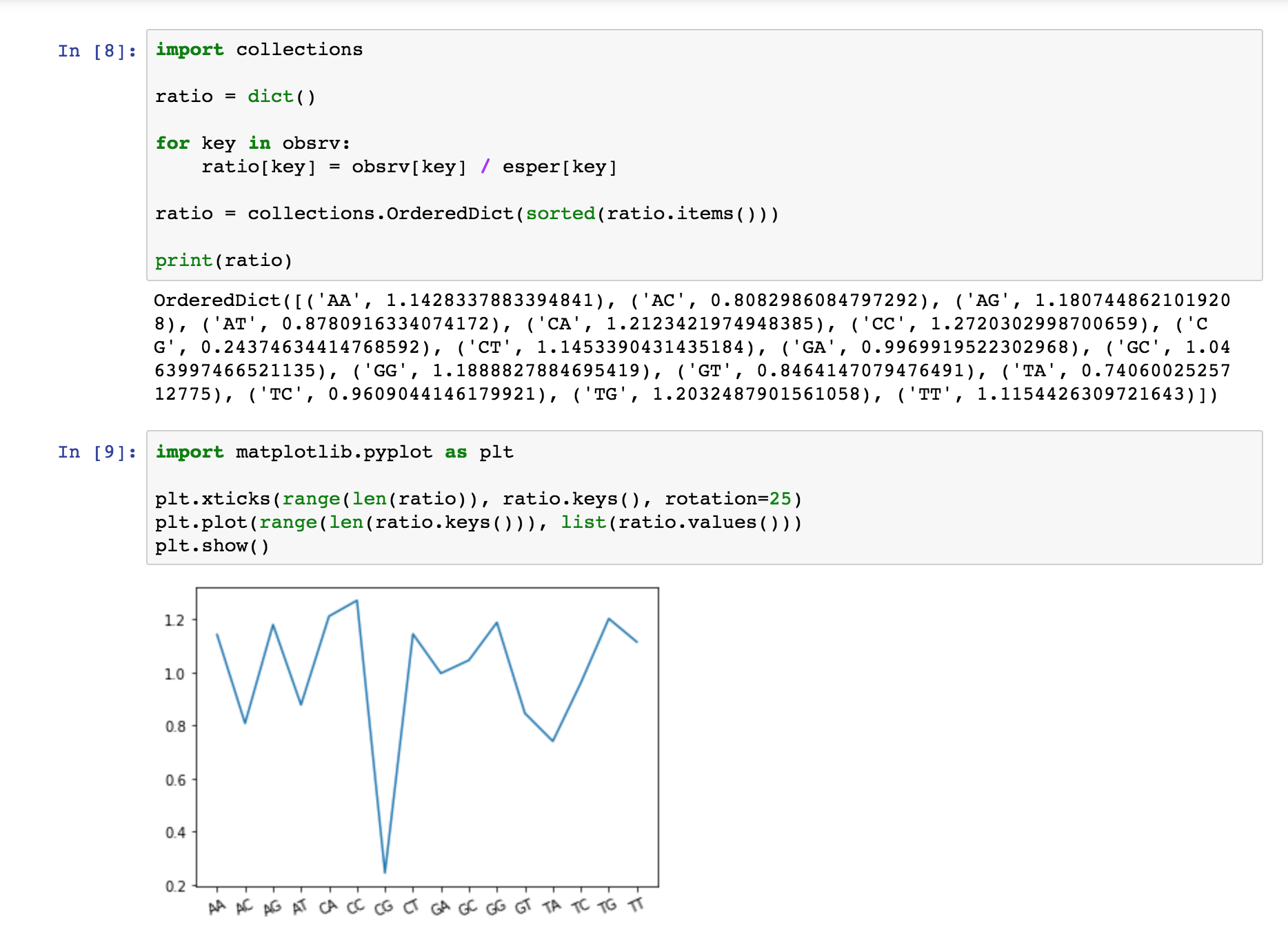
Por último es interesante calcular y representar el ratio observados/esperados de la muestra. Para ello usamos el siguiente código:
ratio = dict()
for key in obsrv:
ratio[key] = obsrv[key] / esper[key]
ratio = collections.OrderedDict(sorted(ratio.items()))
print(ratio)
import matplotlib.pyplot as plt plt.xticks(range(len(ratio)), ratio.keys(), rotation=25) plt.plot(range(len(ratio.keys())), list(ratio.values())) plt.show()
Como podemos observar, el bucle simplemente recorre los diccionarios por cada uno de los tipos de dinucleótidos y divide el número de dinucleótidos observados por el de esperados. Realizamos un plot de la misma forma que para obtener un histograma. En este caso, en vez de usar plt.bar para generar barras, usamos plt.plot para dibujar la gráfica.
Obtenemos así nuestro resultado final: