Introducción
Un microRNA es una molécula de RNA monocatenario implicada en la regulación postranscripcional de la expresión génica. El microRNA maduro tiene una longitud de entre 20-25 nt que se obtiene a partir de la maduración de un transcrito primario (pri-miRNA) más largo. La función de un microRNA depende de la complementariedad entre su secuencia y la secuencia de los transcritos que regula.
Los microRNAs se descubrieron “por casualidad” hace aproximadamente 25 años estudiando el gen lin-14 en Caenorhabditis elegans (Lee, Feinbaum and Ambros, 1993). Estos autores encontraron que la abundancia de la proteína de lin-14 depende de un RNA corto que se transcribe de otro gen, lin-4. La 3’UTR de lin-14 tiene una región parcialmente complementaria a la secuencia madura de lin-4, causando la inhibición de la traducción a la proteína mediante la interacción antisentido (antisense) RNA-RNA. Al principio se pensaba que se podía tratar de un fenómeno específico de nematodos. Sin embargo, durante la última década se han encontrado microRNAs en prácticamente todas las especies de metazoos y plantas, estando muchos de ellos altamente conservados (Axtell, Westholm and Lai, 2011).
Determinar los perfiles de expresión mediante secuenciación masiva
Para detectar microRNAs y determinar sus niveles de expresión, antes de la aparición de los métodos de secuenciación masiva se empleaban técnicas como northern blotting (Lagos-Quintana et al., 2001) o clonación/secuenciación (Landgraf et al., 2007) que no permitían caracterizar todos los microRNAs presentes en la célula de forma simultánea. La aparición de protocolos de secuenciación masiva para los RNAs pequeños ha cambiado drásticamente este escenario, brindando ahora no solamente la posibilidad de medir la expresión de todos los RNAs pequeños a bajo coste, sino también predecir nuevos microRNAs con una sensibilidad y especificidad mucho mayor en comparación con las predicciones in silico (Li et al., 2010).
Los datos de secuenciación masiva del RNA pequeño permiten hasta 4 tipos de análisis diferentes:
- detectar los niveles de expresión de los microRNAs conocidos (por norma general, aquellos que están en la base de datos de referencia miRBase (Griffiths-Jones et al., 2006)),
- detectar aquellos microRNAs que muestran expresión diferencial entre dos grupos (por ejemplo enfermos/sanos, tratados/no-tratados, etc.),
- predecir nuevos microRNAs,
- determinar la presencia de otros tipos de RNAs pequeños.
Para llevar a cabo estos análisis se han desarrollado una serie de aplicaciones que se distinguen notablemente en el número de análisis disponibles y en los métodos empleados. Los programas más usados son: DSAP (Huang et al., 2010) miRanalyzer (Hackenberg et al., 2009; Hackenberg, Rodríguez-Ezpeleta and Aransay, 2011), sRNAbench (Barturen et al., 2014), miRDeep2 (Friedlander, et al., 2012), SeqBuster (Pantano, Estivill and Martí, 2010) y UEA sRNA toolkit (Stocks et al., 2012).
Aunque todos estos métodos pueden diferenciarse en los detalles, comparten algunos pasos:
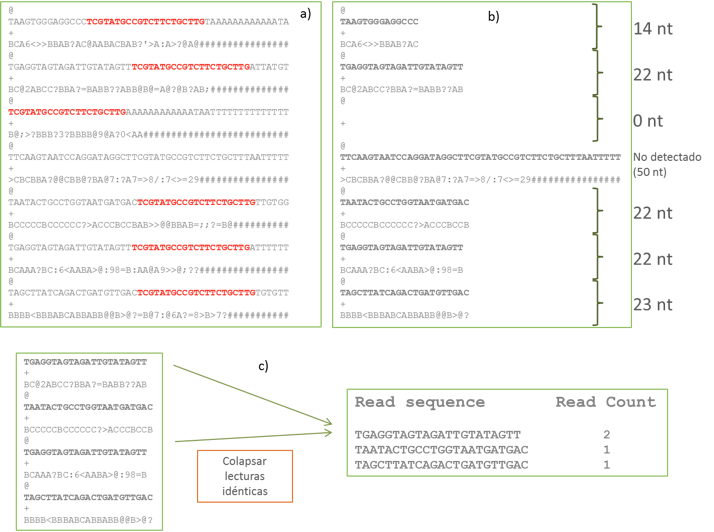
- Preprocesado de las lecturas (véase Ilustración arriba)
- Eliminación de los adaptadores (las lecturas contienen en su extremo 3’ el adaptador que se usa en la secuenciación ya que las lecturas son más largas que las moléculas que se analizan (entre 20 y 30 nt)
- Determinación de la calidad de secuenciación y eliminación de lecturas con baja calidad
- Colapsar las lecturas redundantes, almacenando solamente su secuencia y el número de veces que han sido secuenciadas (read count)
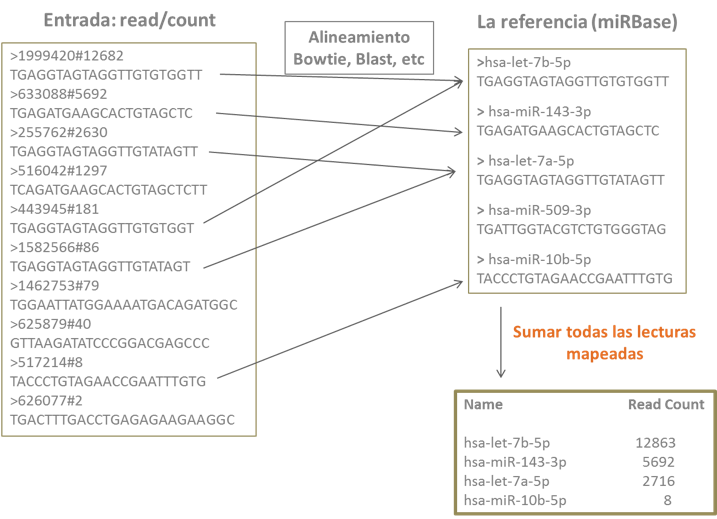
- Asignación de las lecturas (véase Ilustración abajo)
- Mapear todas las lecturas frente a una referencia (generalmente los microRNAs de miRBase)
- Sumar todas las lecturas que mapean a una referencia – este número es directamente proporcional a su nivel de expresión
Aparte de estas dos etapas en común existen otros pasos aguas abajo que si pueden introducir más diferencias
- Tratamiento de alineamientos múltiples: un gran problema en este tipo de análisis consiste en el adecuado tratamiento de lecturas que mapean frente a más de una referencia. Existen tres posibilidades de tomar este hecho en cuenta, pero no está del todo claro cuál es la mejor
- Asignar el conteo (read count) completo a todas las referencias, lo cual sobreestima la expresión total
- Asignar la fracción proporcional a cada referencia, es decir si una lectura tiene conteo 30 y mapea a 3 referencias, se asignaría 30/3=10 a cada una, lo que puede subestimar la expresión de la secuencia de referencia de la que realmente proviene la lectura
- Asignar el conteo completo solamente a una de las referencias siguiendo principios probabilísticos.
- Normalización de las lecturas: este paso es especialmente importante si queremos comparar los perfiles de expresión entre diferentes muestras. No todas las muestras tendrán exactamente, y a veces ni aproximadamente, el mismo número total de lecturas. Pero es obvio, que un microRNA en una muestra con un número alto de lecturas tendrá un conteo más alto que el mismo microRNA en una muestra con menos lecturas. Para tomar en cuenta estas fluctuaciones en el rendimiento hay que normalizar el conteo de las secuencias de referencia. La manera más fácil consiste en calcular el número de lecturas por millón (RPM). Para este cálculo se normaliza mediante el número total de lecturas, siendo no del todo claro si el total debería ser el número de lecturas mapeados frente a los microRNAs o el total de lecturas en el análisis.
- Expresión diferencial: existen varios métodos para determinar los microRNAs que se expresan diferencialmente siendo DESeq (Anders and Huber, 2010) y edgeR (Robinson, McCarthy and Smyth, 2010) los más usados. Los métodos para detectar expresión diferencial pueden sin embargo producir resultados muy diferentes (Seyednasrollah, Laiho and Elo, 2015).