En esta práctica aprenderemos a representar gráficamente datos de expresión utilizando el lenguaje de programación R.
Preparar el entorno de trabajo
Abra una terminal en el servidor de docencia y ejecute R. Verá un mensaje como el siguiente:
R version 3.4.4 (2018-03-15) -- "Someone to Lean On" Copyright (C) 2018 The R Foundation for Statistical Computing ...
Está en una sesión interactiva del interprete de R. Ejecute las siguientes sentencias:
library(pheatmap)
data <- read.delim("/home/biocomp/practica4/TEP_Matrix.tab",
header=T, row.names="GeneSymbol")
Ahora su entorno de trabajo está listo.
Explorar la matriz de expresión
El objeto data contiene la matriz de expresión que utilizaremos en esta práctica. Esta matriz procede de un estudio de mRNA en plaquetas en pacientes control vs. pacientes con cáncer de pulmón de células no pequeñas (el artículo está disponible aquí).
Para ver las primeras 10 líneas de la matriz y las últimas 10 líneas de la matriz ejecute:
head(data, 10) tail(data, 10)
Las filas de esta matriz corresponden a los genes y las filas a los pacientes. Cada valor numérico representa la cantidad relativa de mRNA de un gen concreto en un paciente concreto.
Para obtener una lista de todos los genes ejecute:
genes <- rownames(data) genes
Para obtener una lista de todos los pacientes ejecute:
patients <- colnames(data) patients
Para obtener la cantidad relativa de mRNA de un determinado gen (ej. “DPM1”) en un determinado paciente (ej. “Control_56”) ejecute:
data["DPM1", "Control_56"]
Para obtener la cantidad relativa de mRNA de un determinado gen (ej. “DPM1”) en todos los pacientes ejecute:
DMP1 <- as.numeric(data["DPM1",]) DMP1
Para obtener la media y la desviación típica de la cantidad relativa de mRNA del gen DMP1 ejecute:
mean(DMP1) sd(DMP1)
Para obtener la cantidad relativa de mRNA de todos los genes de un determinado paciente (ej. “Control_56”) ejecute:
Control_56 <- as.numeric(data[,"Control_56"]) Control_56
Obtener un muestreo aleatorio de la matriz
La matriz contiene 3053 genes y 779 pacientes. Compruébelo ejecutando dim(data).
Para facilitar la visualización de la matriz, obtenga una matriz más pequeñas (30 genes aleatorios y 15 pacientes aleatorios) ejecutando:
genes <- rownames(data) patients <- colnames(data) random_genes <- sample(genes, 30) random_patients <- sample(patients, 15) random_data <- data[random_genes, random_patients] random_data
Su matriz aleatoria está en el objeto random_data.
Calcular el Z-score
Aplicaremos una normalización a la escala de color del mapa de calor. Para ello transformaremos las cantidades relativas de mRNA en valores de Z-score.
Ejecute:
cal_z_score <- function(x) { (x - mean(x)) / sd(x) }
random_data_norm <- t(apply(random_data, 1, cal_z_score))
Visualizar la matriz
A continuación vamos a visualizar la matriz contenida en el objeto radom_data. Para ello utilizaremos el paquete pheatmap, que tomará los datos de la matriz y la representará en forma de mapa de calor. Además, pheatmap agrupará los genes y los pacientes en función de su parecido en las cantidades relativas de mRNA. Estas agrupaciones se representan en forma de dendrogramas en los márgenes superior e izquierdo del mapa de calor.
Ejecute:
pdf("my_heatmap.pdf")
pheatmap(random_data_norm)
dev.off()
Y transfiera el fichero my_heatmap.pdf a su ordenador para poder visualizarlo. Debe obtener algo como esto (pero con diferentes genes y pacientes):
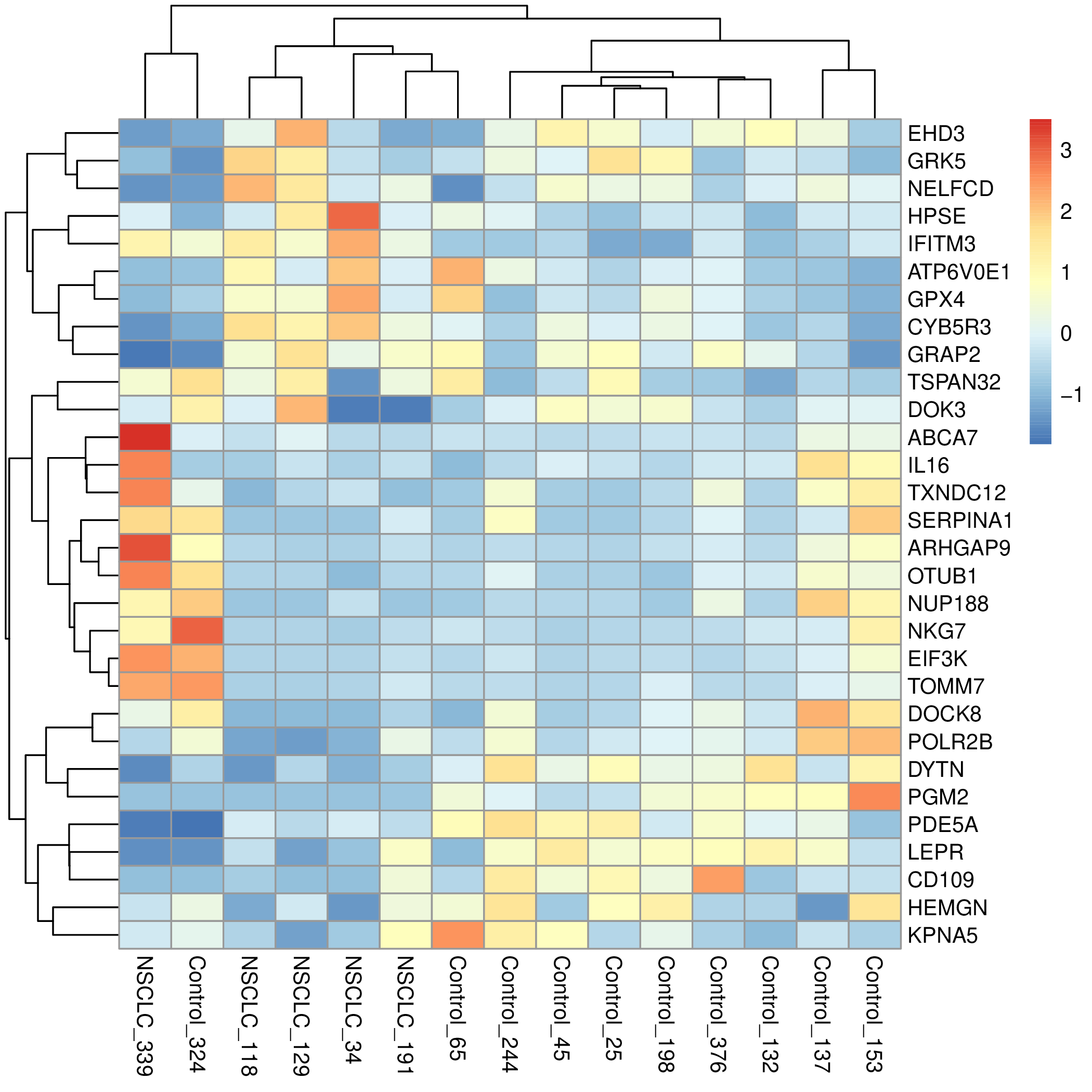
Para facilitar la interpretación del mapa de calor añadiremos una anotación con el tipo de paciente en función de su diagnóstico clínico.
Ejecute:
diagnosis <- read.delim("/home/biocomp/practica4/diagnosis.txt", header=F, row.names=1)
colnames(diagnosis) <- c("diagnosis")
pdf("my_heatmap_diagnosis.pdf")
pheatmap(random_data_norm, annotation_col = diagnosis)
dev.off()
Y transfiera el fichero my_heatmap_diagnosis.pdf a su ordenador para poder visualizarlo. Debe obtener algo como esto (pero con diferentes genes y pacientes):
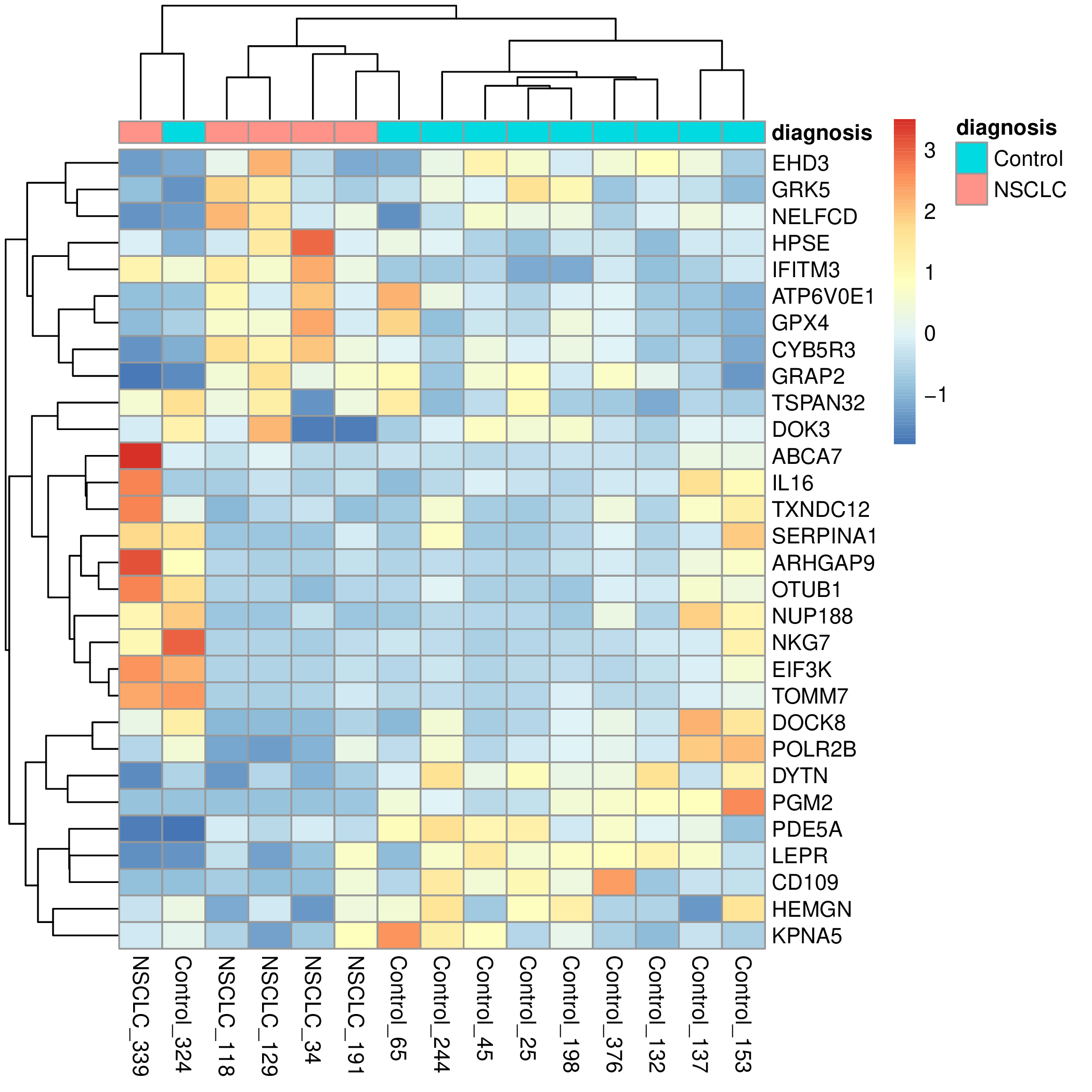
Ejercicios
Seleccionar múltiples genes o pacientes
La función c() de R permite crear vectores, que se pueden utilizar para obtener datos de más de una fila y/o más de una columna de una matriz en una sola operación. Por ejemplo,
data[c('NOP56', 'ETS2', 'AK6'), c('Control_376', 'NSCLC_72', 'NSCLC_9')]
El código anterior devuelve una matriz de 3×3 con los datos de los genes NOP56, ETS2 y AK6 y de los pacientes Control_376, NSCLC_72 y NSCLC_9.
Obtenga una matriz de 5×5 a partir de su matriz random_data y guárdela en el objeto my_data.
Transponer una matriz
La función t() permite transponer una matriz, es decir, intercambiar filas por columnas.
Obtenga transponga la matriz my_data y guárdela en el objeto boxplot_data.
Representar una matriz como boxplots
Vamos a utilizar la función boxplot() de R para representar la matriz boxplot_data como boxplots. Cada boxplot representa la distribución de la cantidad de mRNA de un determinado gen en los cinco pacientes que seleccionó en el primer ejercicio. Habrá un total de cinco boxplot, tantos como genes seleccionó.
Ejecute:
pdf("my_boxplot.pdf)
boxplot(boxplot_data)
dev.off()
Transfiera el fichero my_boxplot.pdf a su ordenador para poder visualizarlo.
Ejecute la ayuda de la función boxplot():
?boxplot
Utilizando esta ayuda, represente los boxplots horizontalmente.