Un alineamiento múltiple consiste en alinear tres o más secuencias a la vez. El resultado es una ‘pila’ de secuencias alineadas de la que se pueden inferir relaciones evolutivas o funcionales.
Datos de ejemplo
- Preproinsulina
- Preproinsulina (con especies añadidas)
- Insulina (CDS)
- Spike protein
- Secuencias de diferentes familias de Alus
Programas de alineamiento múltiple
Programas para representar filogenias
- Phylodendron – IUBio Indiana
- Newick viewer
- PhyloWidget
- phyloGif
- TreeVector
- iToL
- OneZoom Tree of Life Explorer
Recolección de datos
Como hemos visto, para un alineamiento múltiple se necesitan tres o más secuencias de ADN o proteínas en formato multi-fasta.
Hay dos posibilidades para recolectar estas secuencias:
1. A partir de una secuencia anónima:
MVKPIIAPSI LASDFANLGC ECHKVINAGA DWLHIDVMDG HFVPNITLGQ PIVTSLRRSV
PRPGDASNTE KKPTAFFDCH MMVENPEKWV DDFAKCGADQ FTFHYEATQD PLHLVKLIKS
KGIKAACAIK PGTSVDVLFE LAPHLDMALV MTVEPGFGGQ KFMEDMMPKV ETLRAKFPHL
NIQVDGGLGK ETIPKAAKAG ANVIVAGTSV FTAADPHDVI SFMKEEVSKE LRSRDLLD
En este caso se usa FASTA para obtener las secuencias más relacionadas con esta secuencia problema. Marcaremos aquellas secuencias en las que estemos interesados:
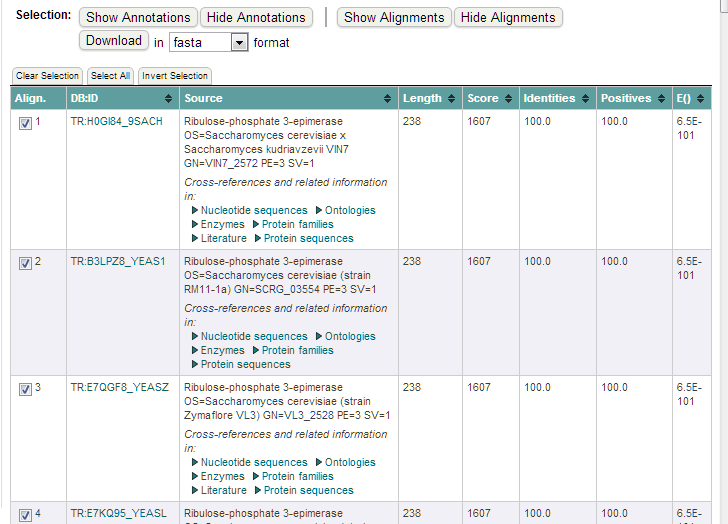
Al pulsar sobre el boton ‘Download’ obtendremos nuestras secuencias en formato multi-fasta:
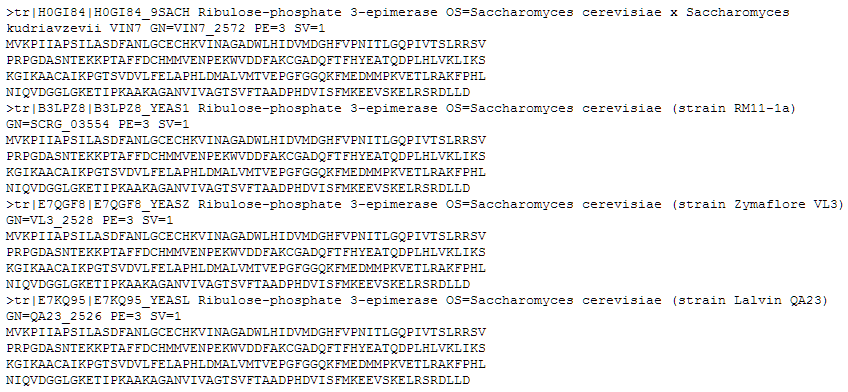
Copiando/pegando este multifasta en el Bloc de Notas, por ejemplo, podremos editar este archivo para poner el nombre adecuado a cada secuencia.
Ejercicio
Se trata de generar una filogenia utilizando alguna proteína que sea de interés: rpe, rpoB, p53, etc.
- Recolecte los datos para hacer un alineamiento múltiple de la manera que se ha visto en el apartado ‘Recolección de datos’.
- Una vez obtenido y formateado adecuadamente el archivo multi-fasta, utilice MUSCLE para obtener el alineamiento múltiple.
- Copie el alineamiento múltiple obtenido, y utilícelo como datos para generar una filogenia mediante ‘ClustalW2 – Filogenia’.
- Recuerde poner en ‘on’ las pestañas ‘Distance correction’ y ‘Exclude Gaps’.
- Copie el arbol resultante (en formato Newick o de paréntesis anidados), y represente la filogenia mediante Phylodendron, PhyloWidget, phyloGif, OneZoom…