Estudio de los sesgos y cuantificación de la metilación del ADN
La conversión de las citosinas no-metiladas por bisulfito y la cuantificación posterior de los niveles de metilación no es una técnica carente de sesgos técnicos. Además, de los sesgos característicos del proceso de secuenciación existen dos sesgos muy conocidos: la conversión incompleta del bisulfito y la interpretación errónea de timinas en las lecturas en caso de la existencia de variantes de secuencia en esa posición. Por ello, para cuantificar correctamente los niveles de metilación deberemos tener en cuenta estos sesgos durante el proceso.
- La herramienta MethylDackel, nos permite por un lado estudiar estos sesgos y tenerlos en cuenta en la cuantificación de los niveles de metilación.
- En primer lugar determinaremos los sesgos por posición durante la conversión por bisulfito:
- Seleccionaremos nuestro alineamiento mediante bwameth
- El ensamblado hg38
- What do you want to do? > Determine the position-dependent methylation bias
- Nos centraremos en la región seleccionada. Region > chr22:30000000-50000000
- Fijaremos la cobertura mínima de secuenciación en 3 lecturas para estimar los niveles de metilación y un mínimo de 5 lecturas para identificar variantes de secuencia. Minimum per-base depth = 3 y Minimum depth for variant avoidance = 5
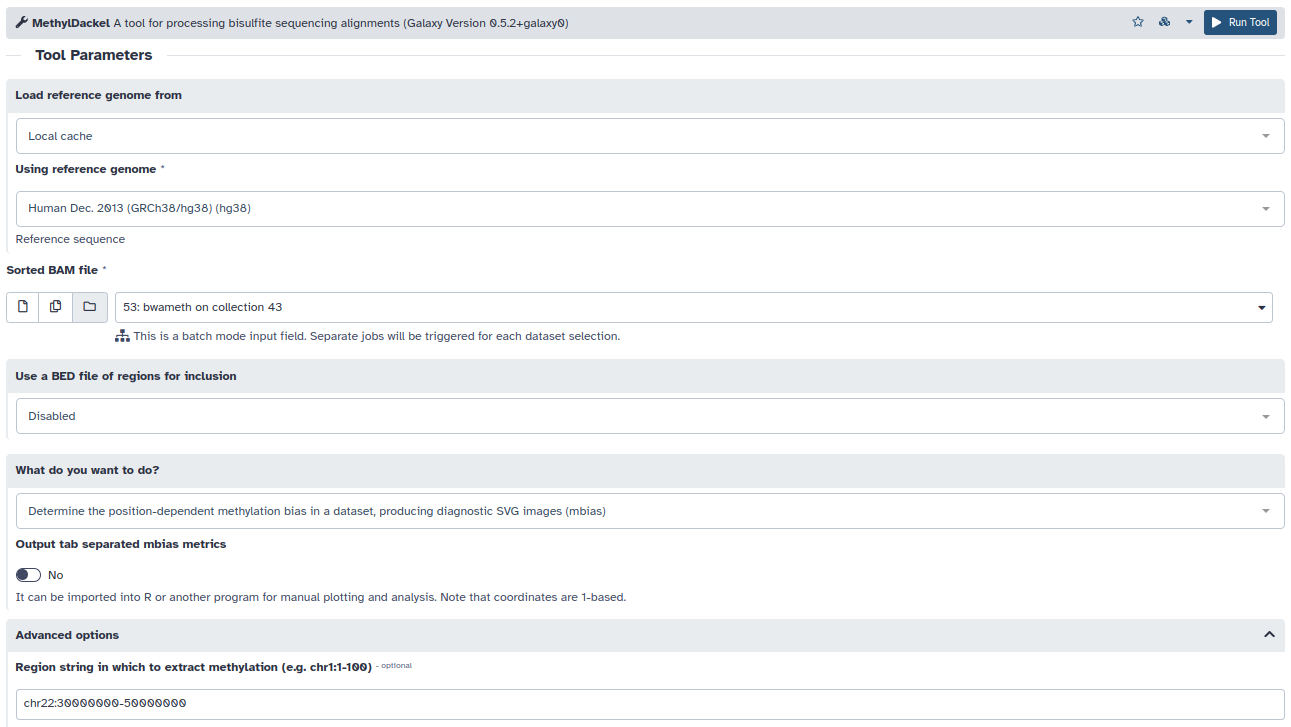
- Debido a la existencia de diferentes protocolos para generar librerías de secuenciación postconversión por bisulfito, la heramienta nos devolverá 4 salidas posibles. Sin embargo, nuestras librerías sólo presentan dos posibles orientaciones: Original top strand y Original bottom strand. Por lo que las otras dos salidas pueden ser eliminadas del historial.
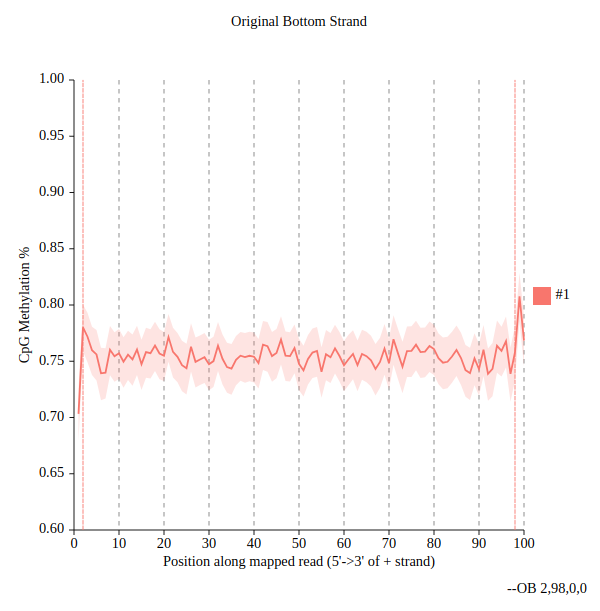
- En el gráfico de la salida podemos observar que la metilación media en las zonas centrales de las lecturas se estima alrededor de 0.75, mientras que los extremos presentan valores ligeramente superiores lo que puede indicar conversiones incompletas por bisulfito en los extremos de las lecturas. Por lo tanto, estos extremos los descartaremos durante la cuantificación de los valores de metilación. Por ejemplo, podemos recortar ambos extremos de las lecturas en 5 nucleótidos lo que nos aseguraría que no tenemos en cuenta este sesgo durante la cuantificación.
- Una vez estimado este sesgo y teniendo en cuenta que MethylDackel tiene en cuenta la presencia de variaciones en la hebra complementaria a la que presenta la citosina (Recordemos que fijamos Minimum depth for variant avoidance = 5) podemos cuantificar con esta misma herramienta la metilación.
- En este caso fijaremos los mismos parámetros que antes salvo:
- What do you want to do? > Extract methylation metrics
- Merge per-Cytosine metrics > Yes
- Los bounds tanto para la Original Top como para la Original Bottom los fijaremos en: 5,95,0,0
- Output options: CpG methylation fractions (valor beta)
- Y en opciones avanzadas recordemos especificar la región y las coberturas mínimas que utilizamos previamente
- Si usamos el ojo en el nuevo objeto del historial, veremos un fichero con formato bedgraph que incluye la localización genómica de cada CpG y su estado de metilación en un valor que oscila entre 0-1
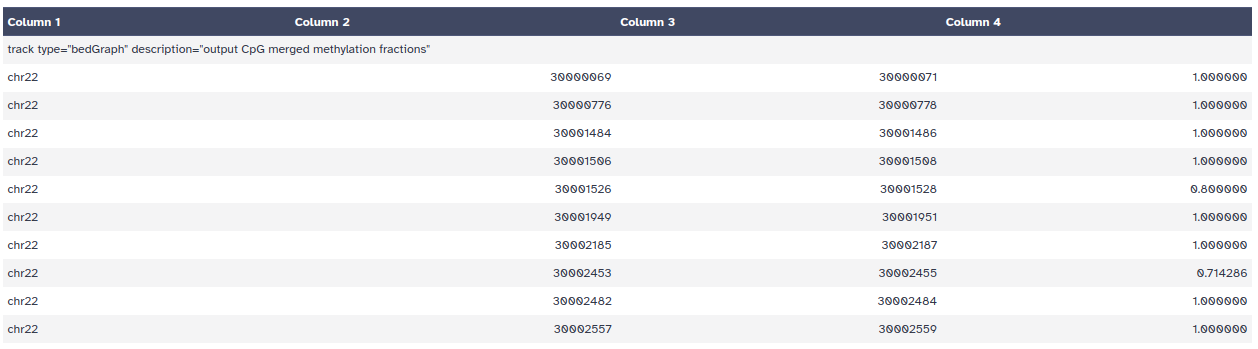
¿Cuál es el estado de metilación de la mayoría de las citosinas? ¿Por qué?
Análisis globales de los patrones de metilación
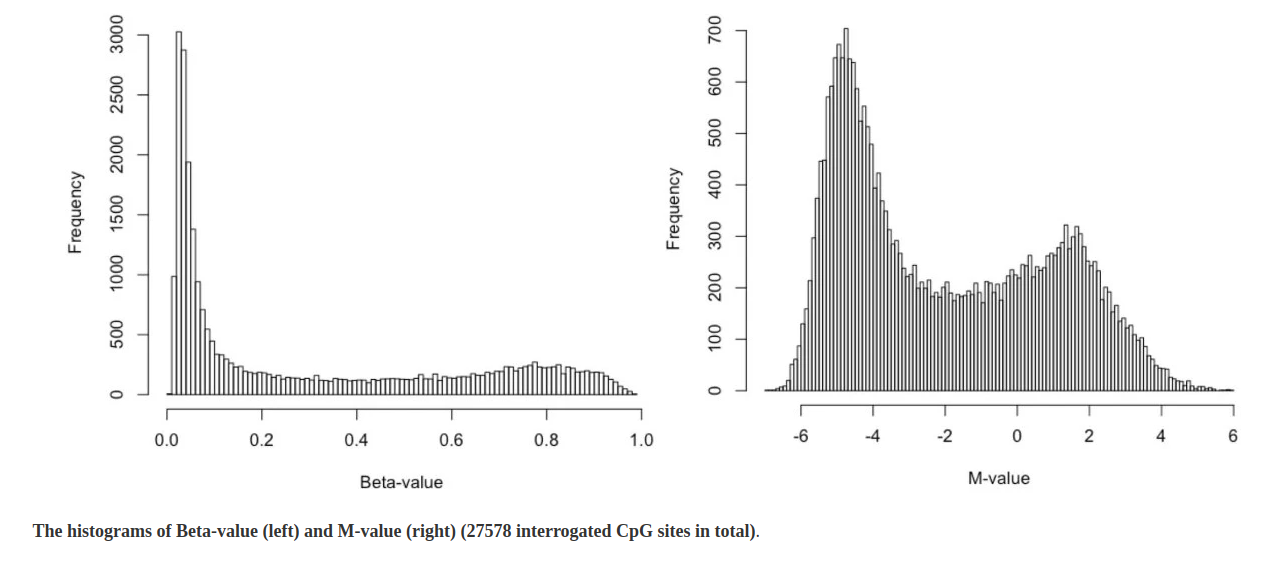
- En primer lugar, estudiaremos la distribución de los valores de metilación. Para ello utilizaremos la herramienta Histogram sobre la 4 columna de la colección de ficheros bedgraphs generados en el paso anterior. ¿Qué observamos en las gráficas resultantes? ¿Qué diferencias existen entre la distribución beta mostrada previamente y las calculadas? ¿Por qué?
- Una vez observadas las distribuciones generales de los valores de metilación, estudiaremos las similitudes entre las muestras. Esto podremos realizarlo mediante correlaciones directas entre las muestras o mediante análisis de reducción dimensional como puede ser el análisis de componentes principales.
- En primer lugar para preparar los datos convertiremos nuestra colección de ficheros bedgraph a ficheros bigWig utilizando la herramienta Wig/BedGraph-to-bigWig. Seleccionad la colección de ficheros bedgraph y corred la herramientas por defecto.
- No todos los CpGs van a tener información para todas las muestras debido a diferentes coberturas por lo que directamente no podemos realizar estudios comparativos entre las muestras. Sin embargo, sabemos que existe cierta similitud en los valores de metilación entre CpGs cercanos, por lo que para solucionar la falta de datos resumiremos los datos en ventanas no-solapantes mediante la herramienta multiBigwigSummary. En esta herramienta seleccionaremos los bigWig que acabamos de obtener, calcularemos las medias en bins (Choose computation mode = Bins), con un tamaño de 100 pbs (Bin size in bp = 100) y especificaremos nuestra región (Region of the genome to limit the operation to = chr22:30000000:50000000). Atención a la separación de coordenadas por dos puntos (:) en lugar de por guión (-).
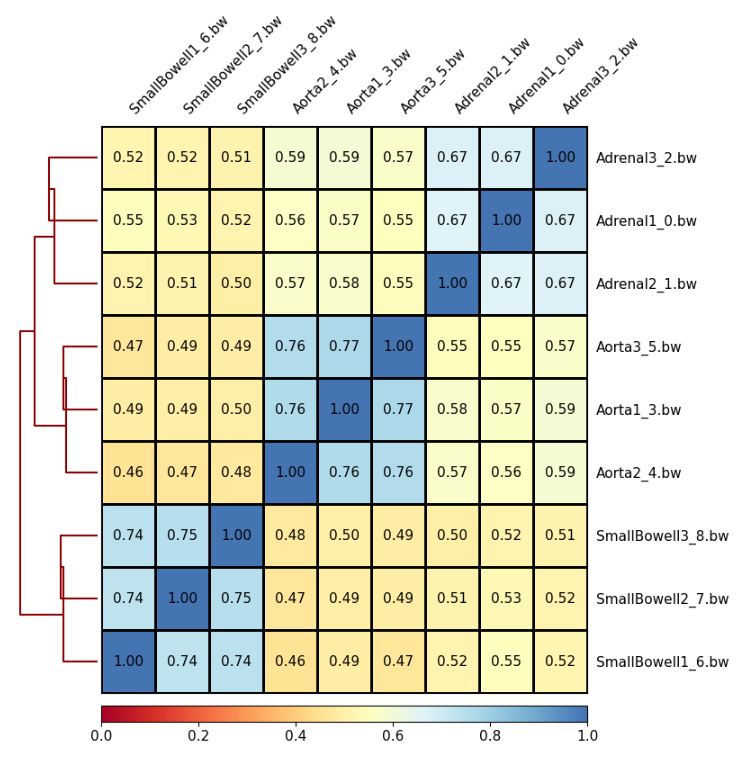
- Una vez resumidos nuestros CpGs en regiones podemos calcular y representar las correlaciones entre las muestras mediante la herramienta plotCorrelation. Aquí podremos calcular correlaciones por pares entre muestras mediante el método de pearson o de spearman, y para visualizar los valores en el resultado activaremos la opción Plot the correlation value. ¿Qué explicación tiene el mapa de calor representado?
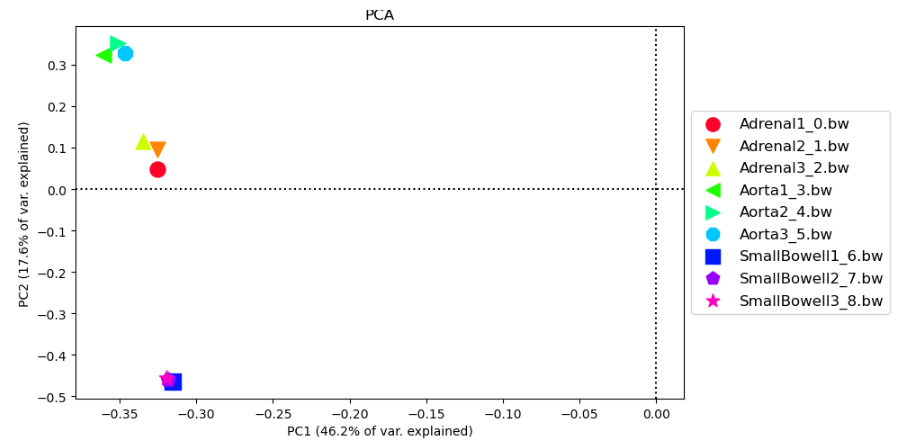
- La relación entre muestras también puede abordarse mediante análisis de componentes principales (PCA), los componentes principales son factores vectoriales ortogonales entre ellos que resumen la variabilidad de los datos. Estos componentes en función de la variabilidad explicada de mayor a menor, permitiéndonos resumir y visualizar en pocas dimensiones la mayor parte de la variabilidad de un conjunto grande de datos. Aquí podéis encontrar un vídeo con una gran explicación de cómo se calculan los componentes principales y cómo interpretarlos.
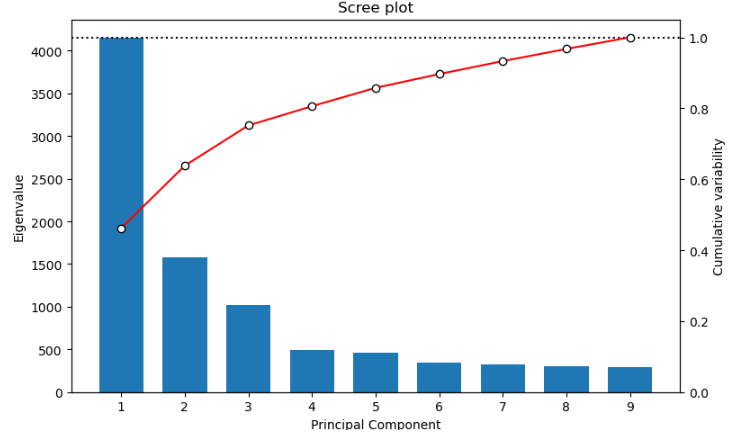
- En nuestro caso utilizaremos la herramiento plotPCA, seleccionad el resumen en ventanas de nuestros niveles de metilación y ejecutadla por defecto, así obtendremos la representación bidimensional de los dos primeros PCs y la varianza explicada por los primeros 9 componentes. ¿Qué observamos? ¿Son similares los resultados a los obtenidos por correlación?