La mayoría de los experimentos en los que se cuantifica la metilación del ADN buscan encontrar diferencias en dichos patrones entre dos condiciones fisiológicas/fisiopatológicas que puedan dar información de los mecanismos moleculares tras dicha condición. Para ello, no es suficiente con comparar las muestras entre dos individuos, ya que los patrones moleculares pueden variar entre individuos o debido a sesgos técnicos. Por lo tanto, para estudiar qué patrones molecular median en el desarrollo de ciertos fenotipos debemos estudiar múltiples muestras de individuos de manera conjunta (cohortes de individuos).
Por otro lado, como vimos en teoría la dinámica del mantenimiento de los patrones metilación provoca que CpGs cercanos presenten valores similares, por lo que el estudio de múltiples CpGs de manera combinada puede permitir obtener resultados más robustos que analizando CpGs individuales.
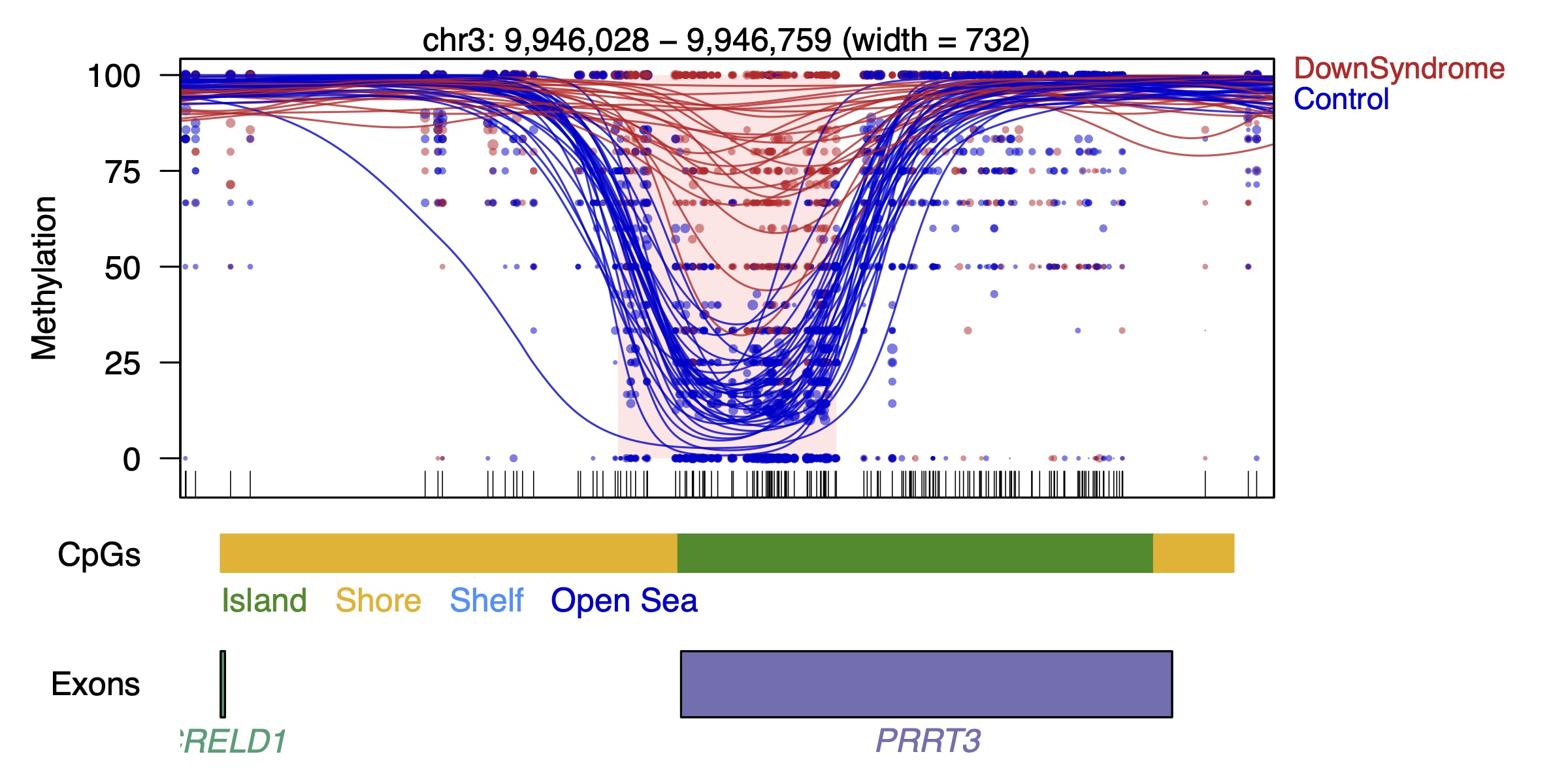
Por último, el análisis de la metilación en el genoma presenta una tercera dimensión. La distribución desigual de CpGs a lo largo del genoma debe ser tenida en cuenta en el análisis, ya que podemos encontrar largas regiones del genoma sin CpGs y por lo tanto sin información de metilación que deben ser tenidas en cuenta en el análisis para definir correctamente las regiones en las que encontramos diferencias funcionales de metilación. Tanto la variación interindividual como la correlación entre CpGs cercanos y la distribución desigual de los mismos puede observarse en la siguiente figura.
Identificación de regiones diferencialmente metiladas
- Los análisis de metilación diferencial se realizan entre pares de grupos de muestras, en nuestro caso seleccionaremos los dos tejidos más dispares basándonos en los resultados de los análisis previos.
- Utilizando la herramienta Filter Collection seleccionaremos las muestras de intestino delgado y de glándula adrenal. Para ello debemos crear dos ficheros de texto con los identificadores de interés (Aorta1, Aorta2, Aorta3 / SmallBowell1, SmallBowell2, SmallBowell3). Estos ficheros los usaremos para filtrar la colección de bedgraphs generados por MethylDackel.
- Input Collection: Colección de ficheros bedgraph generados por MethylDackel (información de CpGs individuales)
- How should the elements to remove be determined?: Remove if identifiers are ABSENT from file
- Filter out identifiers absent from: Fichero con IDs de interés
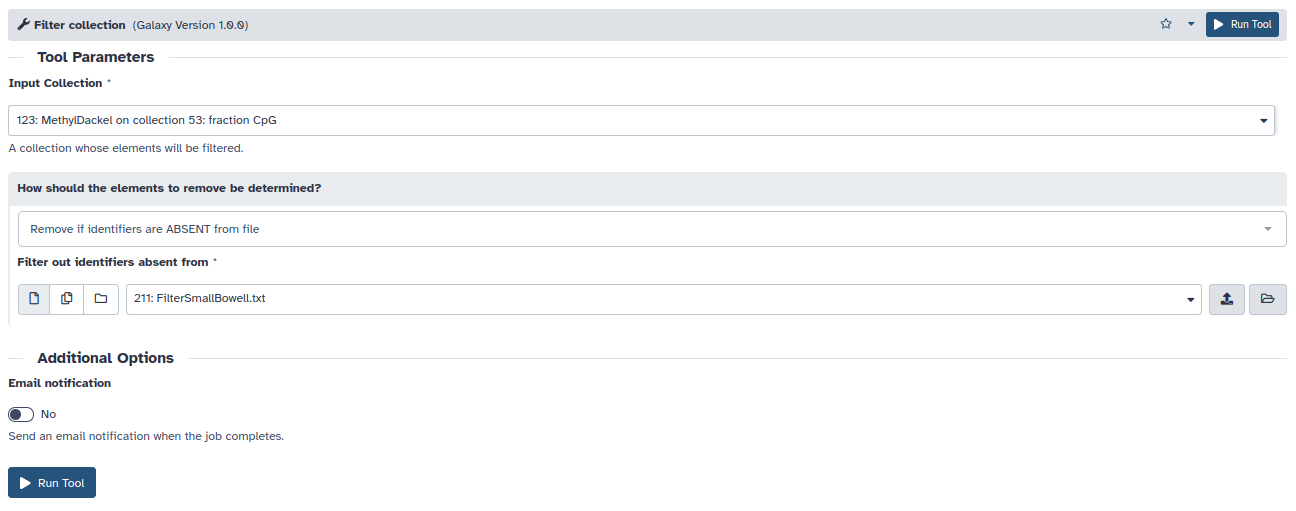
- El filtrado debe realizarse dos veces, una por conjunto de muestras. Filter Collection nos devuelve un objeto con los ficheros seleccionados y otro con los descartados. El objeto con los descartados podemos eliminarlos y mediante el lápiz podemos renombrar ambos objetos seleccionados con las muestras de interés.
- Una de las herramientas que gestiona las 3 dimensiones del problema de la identificación de DMRs (Regiones de Metilación Diferencial) es metilene.
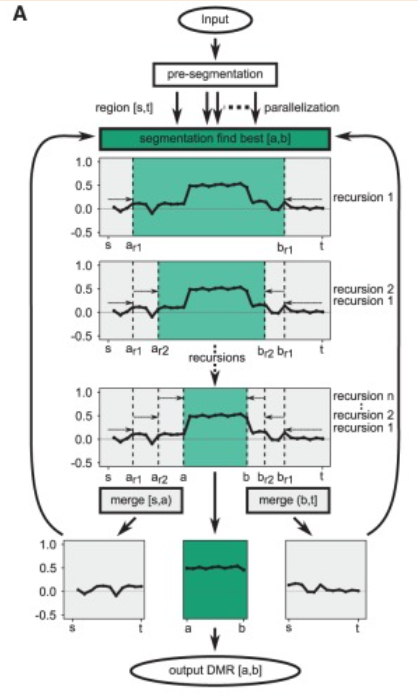
 Metilene se basa en una pre-segmentación para excluir regiones no-informativas, descartando por lo tanto regiones sin CpG y dividiendo el genoma en fragmentos con CpGs localizados a una distancia máxima. Una vez obtenidos los fragmentos a analizar, un algoritmo de segmentación binaria circular analiza recursivamente los valores de metilación entre dos grupos de muestras para identificar regiones que maximicen las diferencias de metilación pero a su vez mantengan valores homogéneos en la región por grupo de muestras.
Metilene se basa en una pre-segmentación para excluir regiones no-informativas, descartando por lo tanto regiones sin CpG y dividiendo el genoma en fragmentos con CpGs localizados a una distancia máxima. Una vez obtenidos los fragmentos a analizar, un algoritmo de segmentación binaria circular analiza recursivamente los valores de metilación entre dos grupos de muestras para identificar regiones que maximicen las diferencias de metilación pero a su vez mantengan valores homogéneos en la región por grupo de muestras.
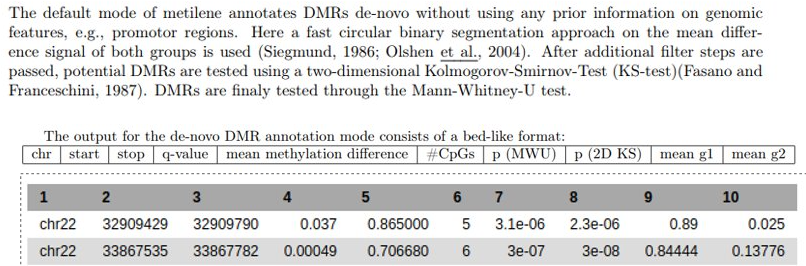
La salida de metilene nos proporciona la localización de las DMRs identificadas y valores estadísticos para estudiar tanto la homogeneidad de la región en cada grupo de muestras (KS) como las diferencias de la región entre los grupos (MWU).
- El algoritmo metilene lo correremos por defecto salvo por el número mínimo de CpGs en una DMR que lo fijaremos en 5 (The minimum # of CpGs in a DMR = 5). Compararemos un conjunto de muestras de un tejido frente al otro, recordad qué tejido habéis incluido segundo, ya que eso marcará la direccionalidad del cambio. En el ejemplo de abajo, Aorta será el primer tejido y por lo tanto los valores diferenciales positivos indicarán aumento de metilación (hipermetilación) en el mismo.
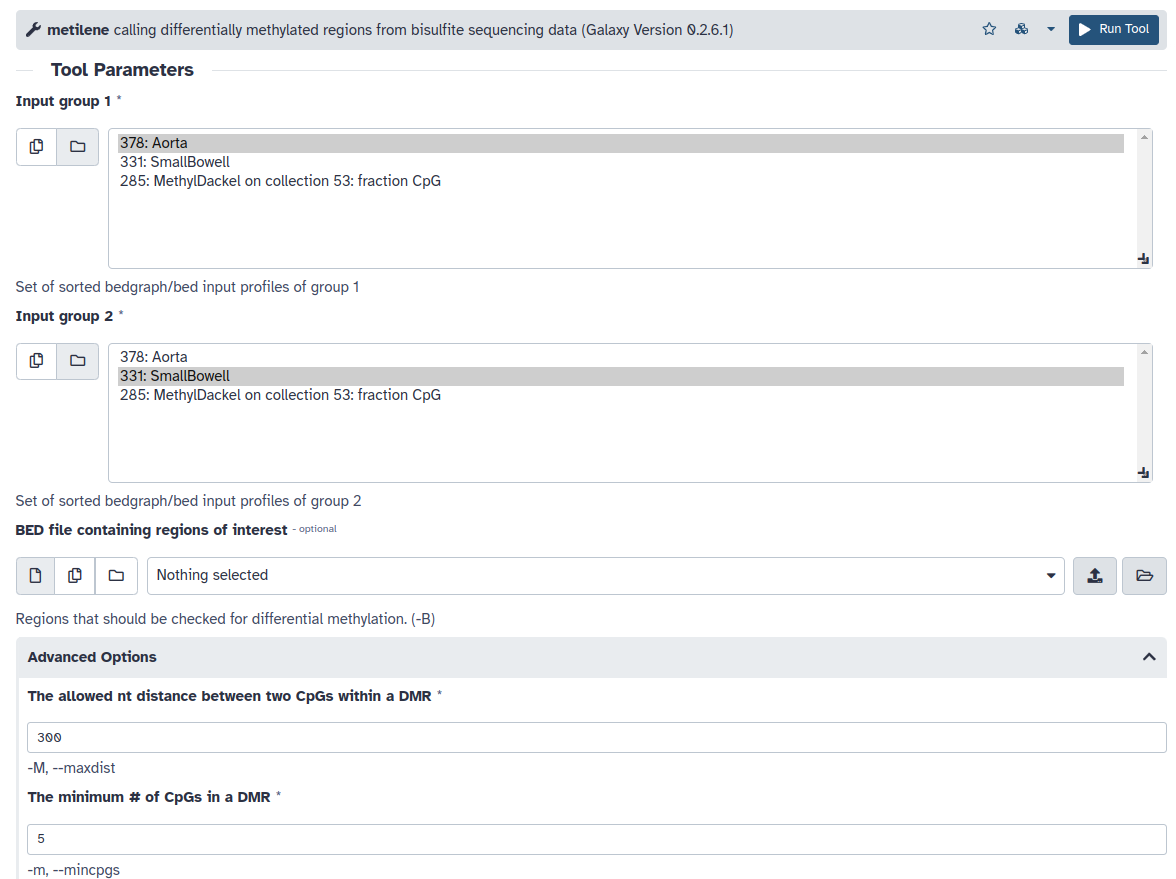
- Nos quedaremos con la primera salida de la herramienta que contiene las posiciones de las DMRs identificadas y los estadísticos de los diferentes análisis. Dividiremos la salida en DMRs entre hipo e hipermetiladas, y seleccionaremos aquellas que presenten diferencias significativas entre grupos y valores homogéneos dentro de cada grupo. Para ello utilizaremos la herramienta Filter data on any column, seleccionando las DMRs con valores de p menores a 0.05 en el test 2D-KS (columna 8) y en el de MWU (columna 7). Este paso lo repetiremos dos veces, una para los valores diferenciales menores de 0 (hipometilados en el grupo de referencia) y otra para valores diferenciales mayores de 0 (hipermetilados en el grupo de referencia).
- c5>0 and c7<0.05 and c8<0.05 (Hipermetiladas)
- c5<0 and c7<0.05 and c8<0.05 (Hipometiladas)
- Una vez separadas las DMRs podemos inspeccionar los cambios de metilación promedios ocurridos en estas regiones. En primer lugar con la herramienta lápiz modificaremos el tipo de datos a formato bed. Fíjese que a pesar de ser un fichero tabulado las 3 primeras columnas corresponden al formato bed: cromosoma, posición inicial y final.
- Mediante la herramienta ComputeMatrix alinearemos nuestras DMRs en el centro y promediaremos los valores individuales de CpGs de los 3 tejidos en esas regiones. Realizaremos este análisis 2 veces, una para las DMRs hipometiladas y otra para las hipermetiladas con los siguientes parámetros.
- Regions to plot: Regiones hipo/hipermetiladas
- Score file: Colección de ficheros bigwig
- computeMatrix has two main output options: reference-point
- The reference point for the plotting: center of region
- Distance in bases to which all regions are going to be fit: 500
- Set distance up- and downstream of the given regions: 500
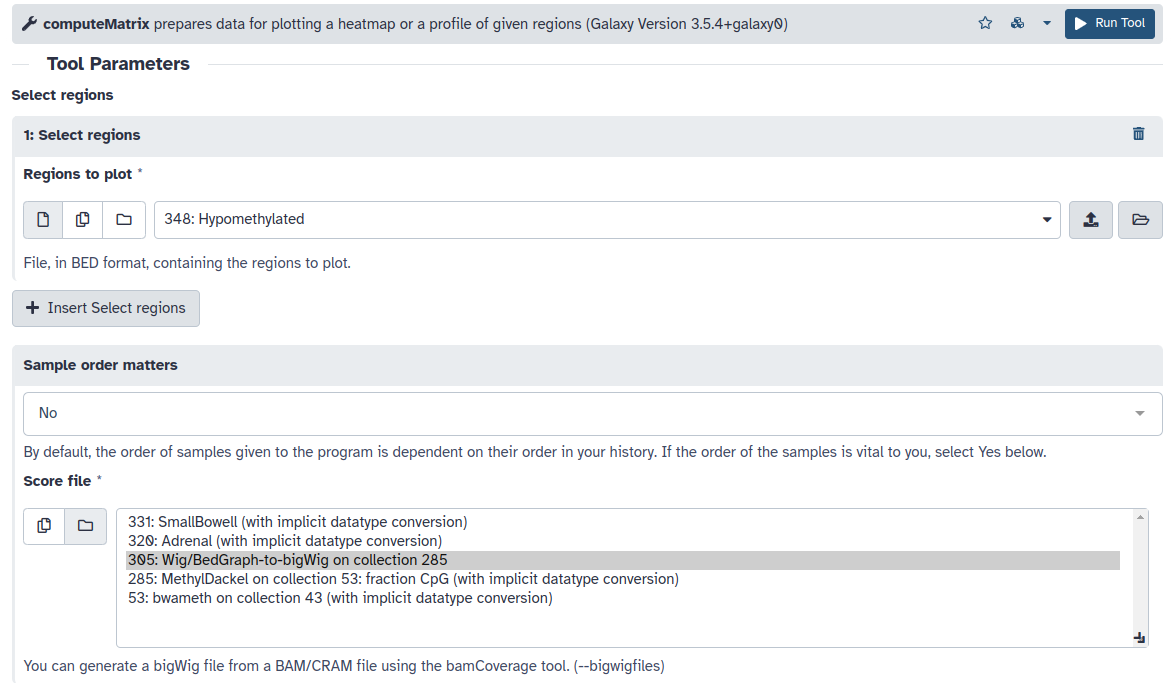
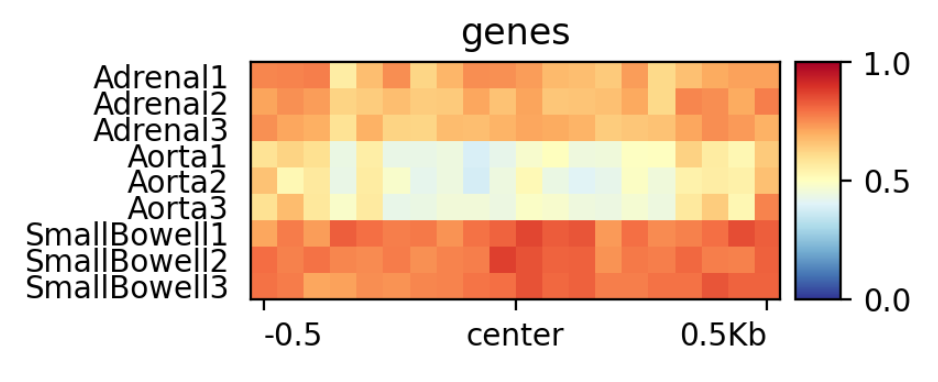
- La visualización la realizaremos mediante plotProfile, en Show advanced options podemos seleccionar el estadístico a representar (Define the type of statistic that should be used for the profile), el tipo de gráfico (Plot type) y agrupar los grupos de muestras en un solo gráfico (Make one plot per group of regions).
Interpretación funcional de las regiones diferencialmente metiladas
- Una manera de analizar qué interacciones pueden estar produciendo los cambios de metilación encontrados puede ser el estudio de TFBS (sitios de unión de factores de transcripción) conocidos en las DMRs. En primer lugar descargaremos la base de datos de TFBSs conocidos para vertebrados desde la base de datos jaspar (PFMs non-redundant y single batch file). Una vez descargado los subiremos a galaxy seleccionando el formato memexml.
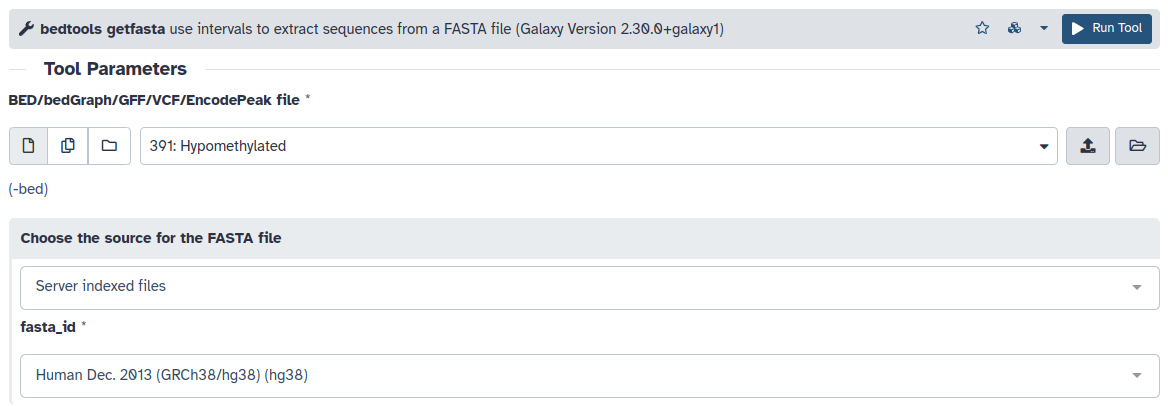
- Después deberemos extraer las secuencias de las DMRs del ensamblado. Utilizaremos bedtools getfasta para a partir de los beds con las DMRs hipo e hipermetiladas extraer las secuencias de ADN del ensamblado hg38.
- Mediante la herramienta FIMO podemos escanear múltiples secuencias en busca de motivos de unión conocidos y estimar el enriquecimiento de estos motivos en las mismas.
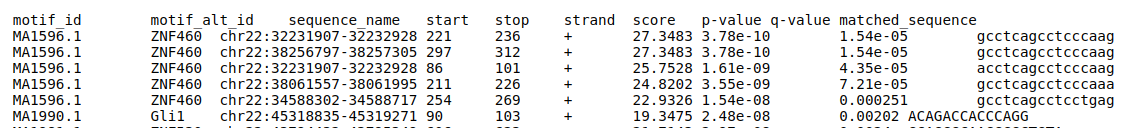
- En el resultado podemos observar qué motivos de unión de qué factores de transcripción están enriquecidos en nuestras secuencias. Estos factores de transcripción pueden tener una importancia funcional y ser los responsables de los cambios de metilación observados.
- Por último, la interpretación de las DMRs suele realizarse por cercanía. Es decir, asociar su función dependiendo de los elementos funcionales o genes que se encuentren más cercanos a cada DMR. Para ello, necesitaremos una anotación génica en formato gtf que podemos subir a galaxy desde este enlace. A este fichero le daremos formato gtf y ensamblado hg38.
- Para cruzar ambas anotaciones los identificadores de referencia deben tener el mismo formato, pero en este caso el gtf no incluye “chr” delante del número de cromosoma. Por lo que modificaremos nuestros ficheros bed de DMRs para adaptarlos mediante la herramienta Replace text in a specific column, en la que reemplazaremos el patrón “chr22” por “22” en la columna 1.
- La herramienta ChipSeeker nos permite anotar por proximidad nuestras DMRs en función de la anotación de genes propuesta. ¿En qué regiones se encuentran preferencialmente las DMRs identificadas?
- Del fichero tabulado con todos los resultados por DMR podemos extraer la lista de genes únicos asociados a nuestras DMRs y realizar análisis de enriquecimiento funcional mediante herramientas tipo EnrichR. Podemos utilizar la herramienta Count occurrences of each record sobre la columna 22 (geneName), copiar los nombres de los genes y utilizar cualquier herramienta en línea. ¿Sería apropiado filtrar ese listado por distancia al TSS? ¿Cómo lo harías?