Introducción
Véase el curso del EMBL-EBI Train online: Human Genetic Variation
Objetivos
El objetivo de este ejercicio es la detección de variantes (SNPs e indels) en ADN genómico extraído de una única persona y enriquecido en exoma, utilizando para ello un pequeño conjunto de lecturas cortas (paired-end) del cromosoma 22 obtenidas mediante NGS en un equipo Illumina GAIIx . Se trata de aproximadamente un millón de lecturas de 76bp (210 Mb en total).
El ADN procede de una mujer caucásica de los EEUU que ha sido resecuenciado multitud de veces, por lo que esta mujer es actualmente uno de los seres humanos mejor caracterizados genéticamente. Su familia fue uno de los 30 trios de los proyectos Hapmap y 1000 Genomas.
El ADN genómico se extrajo a partir de células sanguíneas, enriqueciéndolo a continuación en secuencias del exoma. Para ello, se purificaron utilizando oligonucleótidos específicos en microarrays. Este proceso de enriquecimiento no es perfecto, por lo que en la muestra final puede haber algo de contaminación de ADN no-exómico, asi como ADN mitocondrial.
Filtraremos las variantes obtenidas manualmente, de forma que se comprenda bien el procedimiento. En el mundo real, los conjuntos de datos empleados en la detección de variantes son mucho más grandes y las herramientas bastante más sofisticadas, ya que se basan en modelos estadísticos de la representación de las variantes en el conjunto de las lecturas.
Importación de los datos
- Conéctese a Galaxy y entre en su cuenta (registrese si es necesario).
- En el panel izquierdo de Galaxy pulse Upload File -> Pestaña Collection
- Collection Type: Pair
- File Type: fastqsanger.gz
- Genome: hg38 (2013)
- Paste/Fetch data y pegue los siguientes enlaces en el cuadro de texto que aparece:
- https://bioinfo2.ugr.es/SecuenciasDatasets/ExomeData/NA12878.GAIIx.exome.chr22_1.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/ExomeData/NA12878.GAIIx.exome.chr22_2.fastq.gz
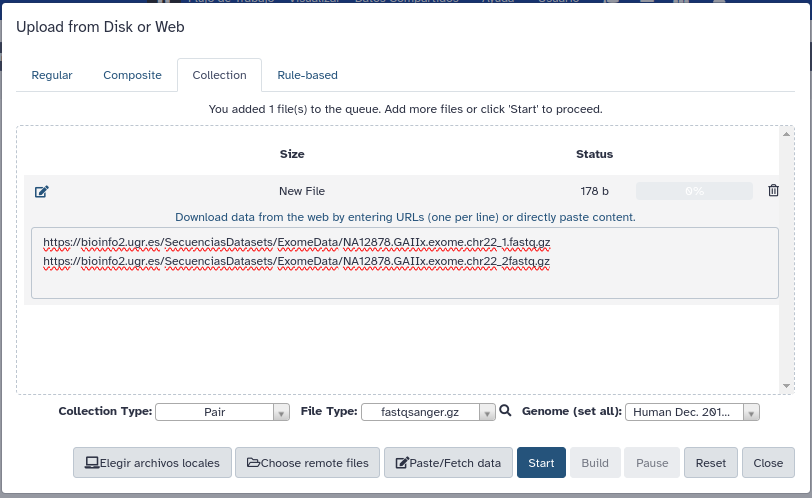
- En el momento de pulsar Start, pulsaremos a su vez la opción Build para agrupar los ficheros. Y crearemos nuestra colección de secuencias emparejadas en el siguiente recuadro.
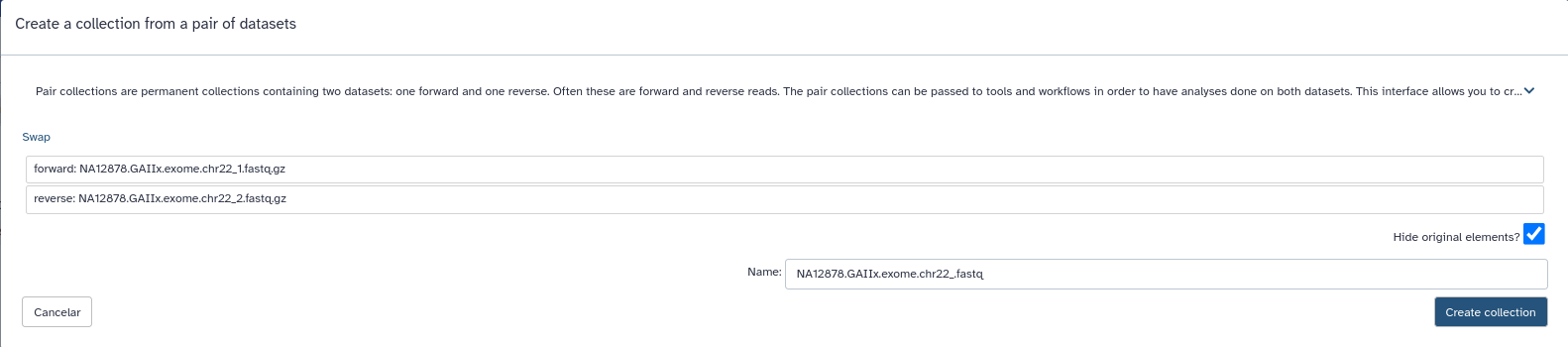
- Cuando acaben de cargarse los datos edítelos con el lápiz del historial y simplifique el nombre.
- A su vez subiremos un fichero con posibles contaminantes del proceso de secuenciación. Para ello volveremos a pulsar Upload File –> Pestaña Regular –> Paste/Fetch data. Pegue el siguiente enlace: https://bioinfo2.ugr.es/SecuenciasDatasets/ExomeData/contaminants_list.txt y seleccione el formato tabular.
- Una vez termine, inspeccione el fichero con el ojo para identificar de donde provienen las posibles secuencias contaminantes durante la secuenciación masiva.
- Asegúrese de que el formato es fastqsanger.gz y el ensamblado hg38.
- Si el formato fuese otro, cámbielo editando los datos mediante el lápiz del historial.
Control de calidad
- Examine las lecturas pinchando en la herramienta OJO.
- Note que en el formato fastq cada lectura está representada por 4 líneas:
-
- Identificador o nombre
- Secuencia
- Separador
- Línea de calidad
- Analice la calidad de las lecturas con FASTQC: En el panel izquierdo de Galaxy, seleccione NGS: QC and manipulation –> FASTQC Read Quality Reports.
- Seleccione como entrada la colección de ficheros fastq pareados que acabamos de generar y como lista de contaminantes el fichero tabulado.
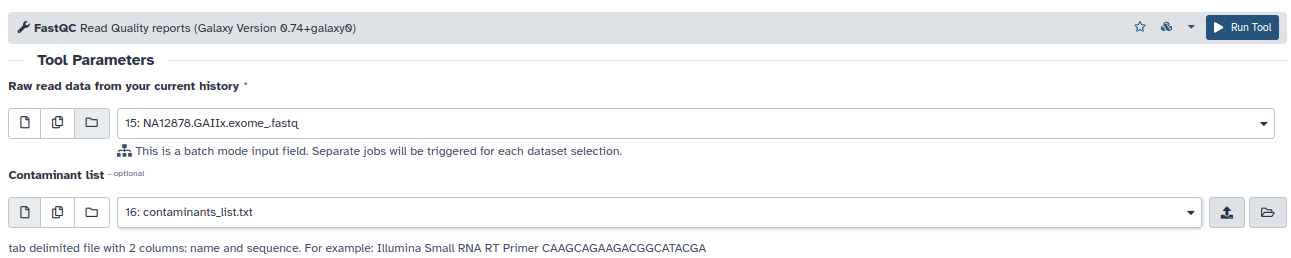
- Deje las demás opciones por defecto y pinche en Execute. Cuando termine el proceso, aparecerán dos items nuevos en su historia, uno con los resultados en forma numérica y otro en html (Webpage). Seleccione este último item y pinche en la herramienta OJO para examinar los resultados (Aquí pueden ver dos ejemplos, uno para datos de buena calidad y otro para datos de mala calidad, que les ayudarán a interpretar sus resultados).
- Aparecerá un FastQC Report conteniendo una lista de gráficos con las medidas de calidad. Examine el gráfico Per base sequence quality. Observe que los datos parecen bastante buenos, la mayoría de los scores están por encima de 30, lo que corresponde a una precisión del 99.9% (véase una explicación de los Phred Quality Scores).
- Note también que el gráfico Sequence Duplication Levels revela que hay cierta tasa de duplicación en las lecturas (artefacto debido a la PCR).
Limpieza de lecturas
- A pesar de que el resumen general de todas las lecturas muestra una buena calidad de secuenciación, siempre puede haber lecturas individuales que mantengan parte del adaptador o presenten una baja calidad de secuenciación en su extremo 3′. Por lo tanto, vamos a proceder a recortar las partes de las lecturas que no nos interesan.
- Utilizaremos la herramienta cutadapt en modo Paired-end Collection y tanto para el Read 1 como para el Read 2 eliminaremos las secuencias en 3′ que contengan los primeros 12 nucleótidos del adaptador universal de Illumina: AGATCGGAAGAG. Por otro lado, antes de buscar los adaptadores eliminaremos los extremos 3′ de las lecturas a partir del nucleótido con un QScore < 20 (Read Modification Option –> Quality Cutoff = 20) y una vez recortadas las lecturas se descartará cualquier par de lecturas con una longitud final menor que la mitad de la longitud original (Filter Options –> Minimum Length = 38).
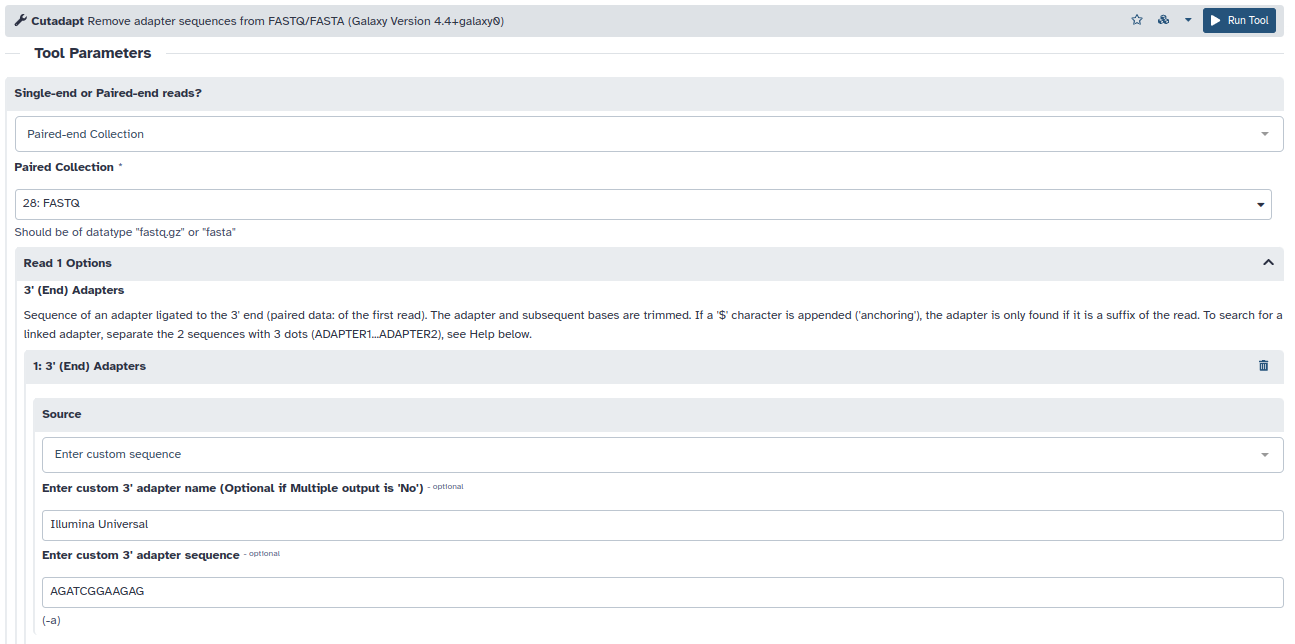
- Una vez seleccionadas estas opciones, ejecutamos la herramienta. El resultado será otro conjunto de lecturas pareadas pero de tamaños variables, para observar los cambios producidos podemos volver a correr la herramienta fastQC sobre la nueva colección. ¿Qué cambios observamos?
Alineamiento y eliminar duplicados
- Usaremos la herramienta Map with BWA para alinear todas las lecturas frente al genoma de referencia. Seleccione nuestros fastq limpios mediante cutadapt y el ensamblado de referencia hg38. El resto de parámetros los dejaremos por defecto y correremos la herramienta.
- Este es el paso que más tarda (5-20 minutos, dependiendo de la carga que tenga el servidor de Galaxy en ese momento).
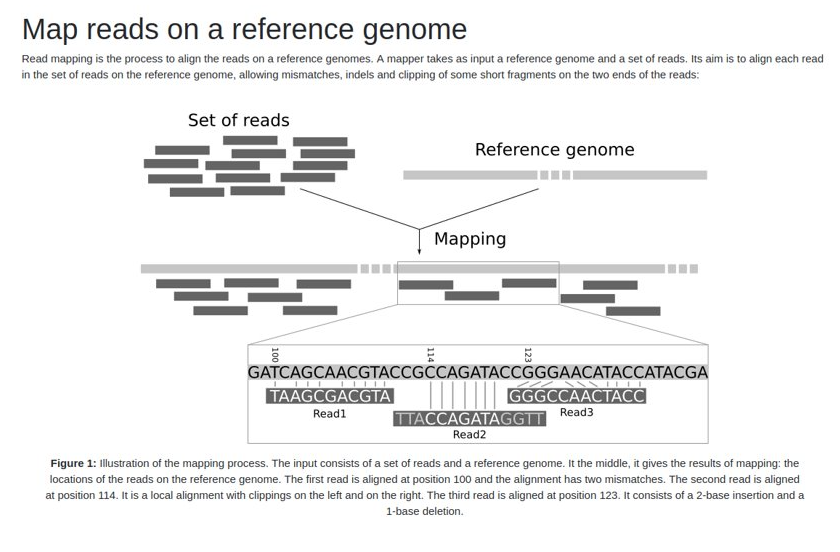
- Cuando termine el proceso, pinche en la herramienta OJO y examine el alineamiento en formato SAM (Sequence Alignment Map).
- ¿Qué es el campo flag de un fichero SAM?
- A partir del alineamiento eliminaremos todas aquellas lecturas cuya posición inicial de alineamiento sea la misma, ya que pueden ser secuencias duplicadas durante la amplificanción en el flowcell. Esto lo realizaremos mediante la herramienta RmDup, indicando que nuestro fichero BAM (binario del SAM) contiene lecturas paired-end.
- Observe que muchas lecturas no mapean en el cromosoma 22 (columna 3). Para quedarnos solamente con aquellas lecturas que mapean correctamente en el cromosoma, debemos filtrar estos resultados samtools view (A filtered/subsampled selection of reads –> Manually specify regions –> chr22). De esta manera, en el nuevo item de su historia, solo aparecerán ahora aquellas lecturas que mapean en el cromosoma 22.
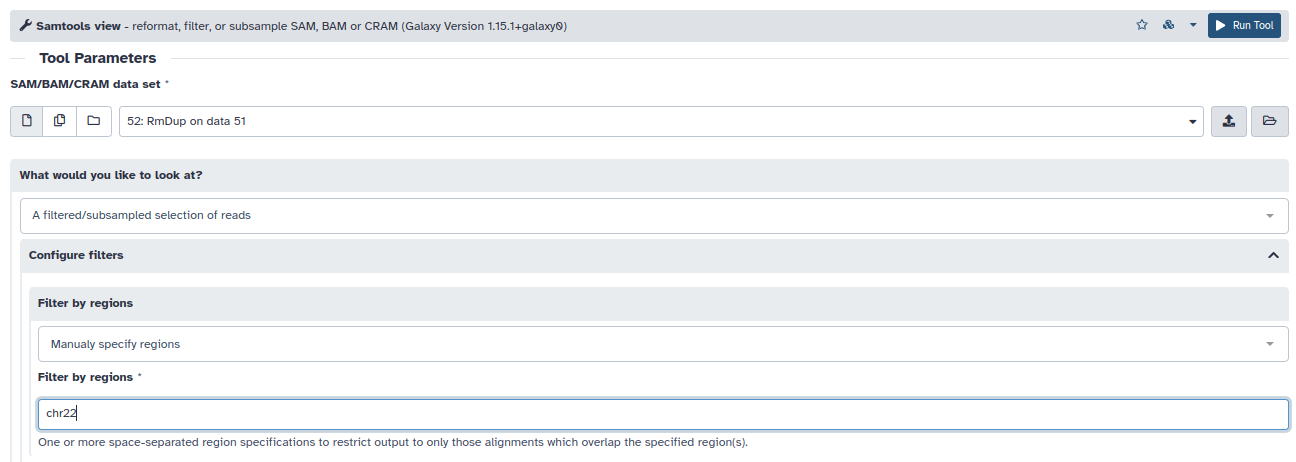
- Con la herramienta lápiz renombre este último item a: BAM_chr22_mapped
Visualización
- Para visualizar el alineamiento en el UCSC Genome Browser, pinche en Visualize y elija display at UCSC main.
- Si activa el track de genes RefSeq y pone las coordenadas chr22:35,393,817-35,431,754 en el cuadro de texto del Genome Browser obtendrá una imagen como esta:
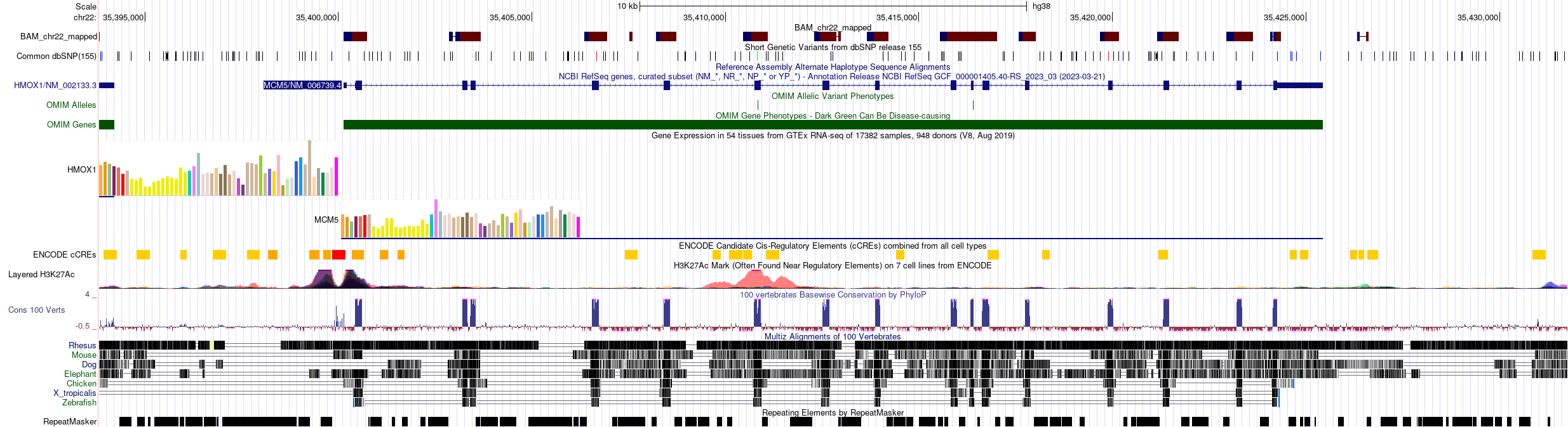
¿Qué observamos en esta imagen? ¿Explora la representación de las lecturas (BAM_chr22_mapped)? ¿Qué significan sus colores?