Filtrado y validación de los SNPs (SNP calling)
- Para obtener información más detallada sobre cada posición del alineamiento, usaremos el programa Generate pileup. Este programa facilita la selección e identificación de los SNPs más relevantes. Ejecute pileup sobre las lecturas alineadas que obtuvo en el ejercicio anterior (SAMtools –> Generate Pileup). Mantenga todas las opciones por defecto, excepto Call consensus according to MAQ model –> Yes. Esto genera una base de consenso en cada posición cromosómica. Este cambio anterior abrirá más opciones; manténgalas todas por defecto y pinche en ‘Execute‘.
- Cambie a formato pileup el archivo que acaba de generar: Herramienta lápiz –> Datatype –> pileup. El archivo pileup resume los datos de las lecturas en aquellas regiones genómicas cubiertas por al menos una lectura.
- Para aumentar la calidad de las variantes obtenidas, aplicaremos dos filtros sucesivos:
- Filter Pileup
- Select ‘Pileup with ten columns (with consensus)’
- Do not report positions with coverage lower than = 10
- Convert coordinates to intervals = Yes
- Filter and Sort –> Filter
- Seleccione los SNPs que tengan ‘c7>50′. La columna 7 contiene un score que combina varias medidas de calidad, como la cobertura o la calidad por posición.
- Filter Pileup
¿Cuántas variantes encontramos tras los filtros utilizados?
Visualización en Genome Browser y validación con dbSNP
- Mediante el pileup filtrado podemos identificar posiciones en homocigosis o heterocigosis y visualizarlas en los alineamientos de las lecturas como los ejemplos siguientes:
Variante en homocigosis anotada en dbSNP:
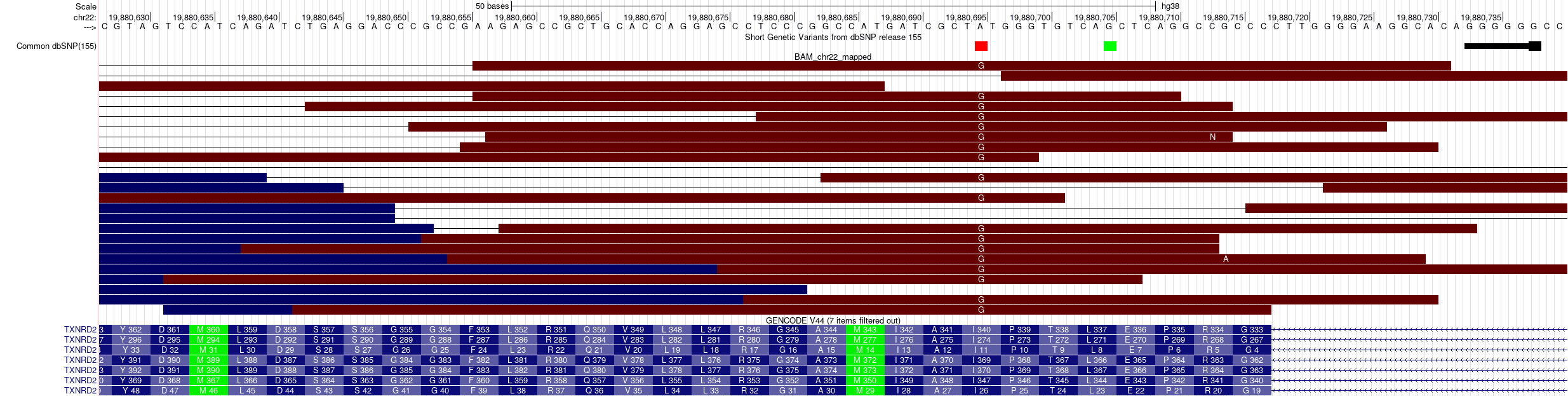
Si hacemos clic en la variante de dbSNP, podemos obtener información adicional de dicha variante.
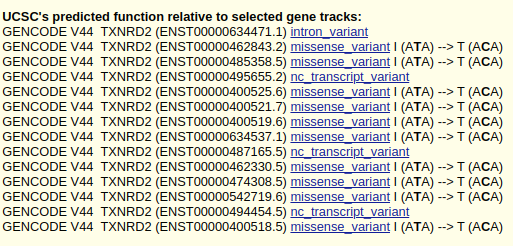
Así pues, se trata de una variante homocigótica A/G que provoca un cambio no-sinónimo en la segunda posición de un codón que codifica para Isoleucina –> Treonina.
Encuentra una variante adicional en heterocigosis e inspecciona la información disponible en dbSNP para la misma.