Algoritmos de predicción
Una vez obtenida una secuencia de interés durante un experimento de biología molecular, es de gran interés identificar posibles marcos abiertos de lectura (ORFs). Estos marcos abiertos de lectura proporcionan información crucial de la posible funcionalidad de la secuencia, ya que su presencia puede suponer que esa secuencia de ADN codifica una proteína. Una vez identificados se puede estimar que dominios funcionales contiene esa proteína y por lo tanto determinar su posible funcionalidad.
Estas predicciones de las presencia de ORFs puede abordarse en base a modelos teóricos de la estructura génica: potencial codificador, secuencias donadoras y aceptoras durante el “splicing” alternativo o la presencia de elementos reguladores como promotores. Este tipo de aproximaciones se denominan “ab-initio”. Por otro lado, si se conocen los genes en especies próximas a la especie en estudio, la predicción de genes puede basarse en la homología de secuencias, aunque generalmente suelen integrase con la información procedente de modelos teóricos (Hybrid approach).
Ejercicios
- Identificar marcos abiertos de lectura y la posible proteína codificada
- A partir de la siguiente secuencia problema identificaremos la posible presencia de ORFs mediante el programa orffinder. Identifica el ORF más largo presente en la secuencia. ¿Cuál es su orientación en la hebra? ¿En qué marco de lectura se encuentra?¿Cuál es su longitud nucleotídica y aminoacídica?
- Para identificar su posible funcionalidad realizaremos una búsqueda mediante el algoritmo blastp de su secuencia aminoacídica. ¿Existe alguna proteína similar a la codificada por el ORF en la base de datos? El gen del que se codifica dicha proteína, ¿Cuántos exones presenta? ¿Presenta algún dominio funcional conocido?
- Predecir la existencia de genes en una secuencia anónima a partir de un modelo ab-initio propio.
- Identifica mediante orffinder los posibles ORFs presentes en la secuencia problema. ¿Cuántos posibles ORFs encontramos? ¿Qué puede estar ocurriendo?
- El programa NetGene2 puede darnos algunas pistas adicionales, introduce la secuencia completa. Inspecciona los resultados. ¿Qué información adicional nos proporciona? ¿Nos hacemos una idea de lo que ocurre?
- Mediante la información proporcionada por ambos programas predice la posible estructura del gen, y anota y verifica sus posibles dominios proteínicos con blastp.
Bondad de predicción (Genes)
La bondad de las predicciones computacionales suelen estimarse en función de:
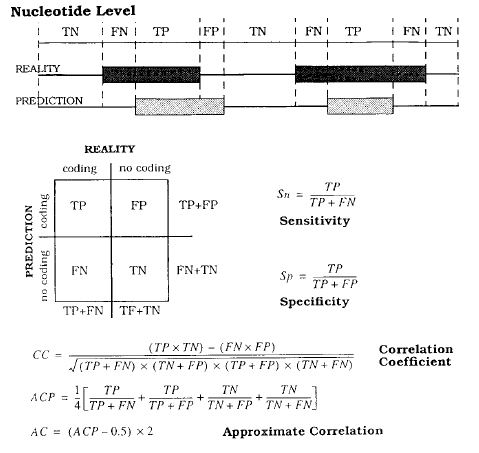
- Sensibilidad: fracción de elementos identificados correctamente por la predicción (Verdaderos positivos, TP) y el total de elementos conocidos (TP + FN, Falsos Negativos o elementos conocidos no predichos). Es decir, la capacidad de detectar los elementos conocidos.
- Especificidad: fracción de regiones que no son elementos identificadas correctamente (Verdaderos negativos, TN) y el total de regiones conocidas que no contienen elementos (TN + FP, Falsos Positivos o regiones sin elementos pero predichas como tal). Es decir, la capacidad de identificar regiones sin elementos conocidos.
En el caso de elementos genómicos, estas medidas suelen cuantificarse en función de los nucleótidos totales. Así un elemento predicho correctamente sumaría el total de su longitud a los TPs. Esta aproximación presenta un gran problema, y es que elementos minoritarios (en términos de longitud) en el genoma presentarían un número elevadísimo de TN, ya que la mayoría de los nucleótidos del genoma no corresponderían con dicho elemento. Por lo tanto, en la fórmula de la especificidad sus valores siempre rondarían el máximo (~1) siendo imposible por lo tanto de discriminar entre predicciones cercanas.
Burset y Guigó en 1996 propusieron una modificación en la especificidad para tratar de ampliar el rango de la misma en las predicciones genómicas. En resumen, propusieron el uso de lo que se conoce como valor positivo de predicción o precisión (PPV = TP / (TP+FP)) en lugar de la especificidad, como se puede observar en la siguiente figura. En este caso en lugar de valuar la capacidad de identificar elementos negativos, se evalúa la fracción de predicciones correctas (TP) sobre el total de predicciones (TP+FP).
Ejercicio
Compararemos la bondad de la predicción de nuestra predicción de genes en la secuencia problema del ejercicio anterior con la predicción obtenida por 2 herramientas adicionales de predicción de genes eucariotas: Genscan (ab initio) y GeneID (hybrid).
- En primer lugar debemos identificar qué genes hay descritos en esa secuencia problema. Por lo que primero deberemos identificar su localización genómica (usando la herramienta BLAT del UCSC por ejemplo), y después utilizando el Table Browser de la UCSC descargaremos las posiciones de los CDS de los genes contenidos en dicha secuencia según el NCBI RefSeq Curated en un nuevo historial de Galaxy (al igual que hicimos en ejercicios anteriores). ¿Qué diferencias observamos entre las coordenadas obtenidas del UCSC y las coordenadas estimadas de la predicción original de los CDS? ¿A qué se debe?
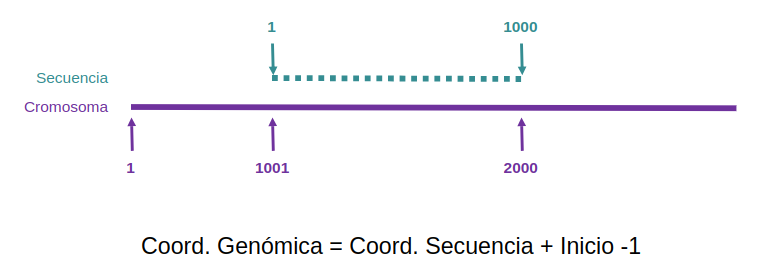
- Debemos corregir las coordenadas de secuencia a coordenadas genómicas para que sean comparables. Una vez corregidas generaremos un fichero con formato BED con las mismas. Las coordenadas de las predicciones están en base 1 y las posiciones genómicas del buscador del UCSC también por lo que hay que ajustarlo a base 0 para poder compararlas con las posiciones de la base de datos del UCSC (en total debe restarse dos a las posiciones inicales y uno a las finales tras el ajuste genómico).
- Usando de nuevo la secuencia problema obtendremos las predicciones de los software Genscan y GeneID, y al igual que antes corregiremos las coordenadas y generaremos sendos ficheros BED con las predicciones.
- Calcularemos los conteos para calcular la bondad con Galaxy, por lo tanto subiremos los ficheros bed generados al historial. Para ello usaremos la función “Upload Data”:

Seleccionaremos los ficheros locales y elegiremos el formato de los ficheros como BED y el ensamblado del genoma utilizado (por ejemplo: hg38).
- Como se mencionó al inicio, la bondad se calcula en base a los nucleótidos predichos y los anotados. Por lo tanto, para calcular los valores de TP, FP, TN y TP utilizaremos funciones que nos calculen las intersecciones entre nuestras predicciones y los CDS anotados. En primer lugar calcularemos los TP mediante la herramienta “intersect” que nos calculará la intersección entre cada una de las predicciones y los CDS anotados.
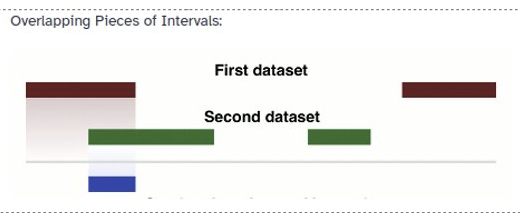


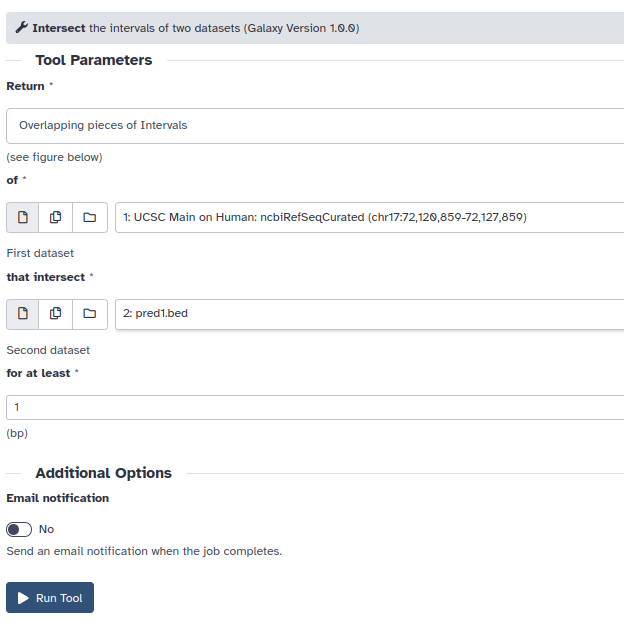
Esta herramienta nos devolverá al historial un item con las regiones solapantes entre ambos ficheros y mediante la herramienta “Summary Statistics” calcularemos la longitud de los mismos, cuya suma será el valor de TPs que anotaremos en una hoja de cálculo.
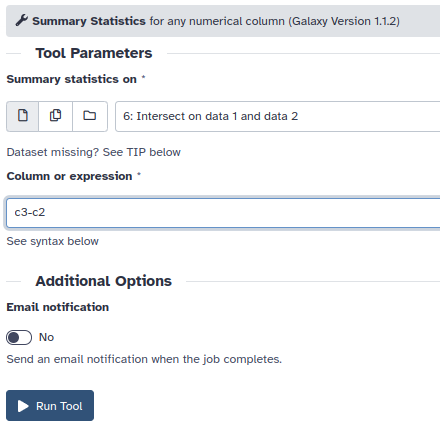
- Ahora calcularemos los FN como la longitud total de los exones anotados – TP previamente calculados. La longitud de los exones se puede calcular al igual que en el punto anterior mediante “Summary Statistics”. Y anotaremos los resultados en la hoja de cálculo junto con los TPs.
- ¿Cómo podemos calcular los FP? ¿Y los TN?
- Una vez obtenidos los valores para las 3 predicciones, calcular Sp, Sn y PPV en base a las fórmulas indicadas arriba y comparar los resultados entre predicciones. ¿Puedes estimar cuál es la mejor predicción?