Objetivos
El objetivo de este ejercicio es cuantificar los valores de metilación del ADN en una región del cromosoma 22 (región 30.000.000 – 50.000.000) de 3 individuos en 3 tejidos diferentes de cada uno (Glándula adrenal, Aorta e Intestino Delgado). Para ello se ha seleccionado un pequeño conjunto de lecturas (single-end) de dicha región del genoma que fueron secuencias tras tratar el ADN con bisulfito.
Durante el ejercicio utilizaremos herramientas fundamentales en el análisis y la interpretación de datos procedentes de la secuenciación con bisulfito para comprender las diferencias frente a la secuenciación sin bisulfito, y qué sesgos específicos deben considerarse en este caso. Además, en este ejercicio observaremos la relación tejido-específica que presenta la epigenética a diferencia de la genética cuyas diferencias son individuos-específicas.
Recomendación importante: Durante este ejercicio se generarán numerosos ficheros, así que se recomienda ir renombrándolos para saber en todo momento qué contienen.
Importación de los datos
- Al igual que durante la identificación de variantes, mediante la opción Upload Data subiremos los ficheros problema desde la pestaña Collection con Paste/Fetch usando los siguiente enlaces:
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Adrenal_1f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Adrenal_2f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Adrenal_3f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Aorta_1f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Aorta_2f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/Aorta_3f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/SmallBowell_1f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/SmallBowell_2f.fastq.gz
- https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/SmallBowell_3f.fastq.gz
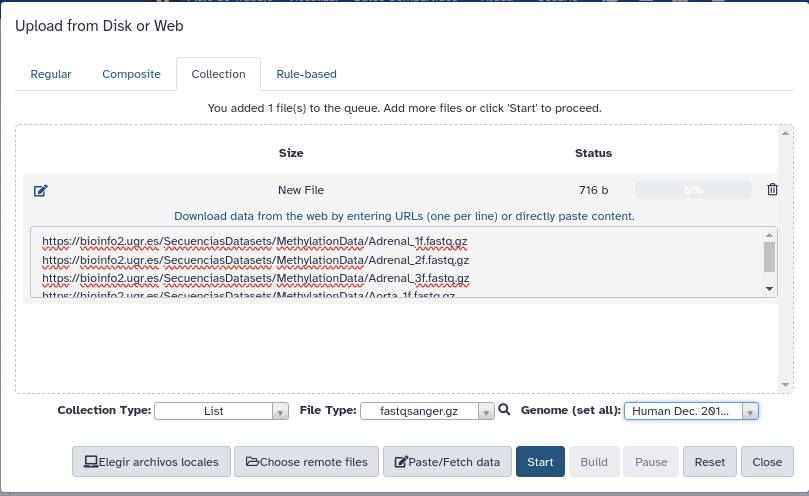
- Aseguraros de especificar las opciones Collection Type: List, File Type: fastqsanger.gz y Genome: hg38 (2013), y una vez pulsado Start deberéis pulsar también Build para generar la colección de los fastq y poder trabajar conjuntamente con todos a la vez.
- Ahora subiremos la lista de contaminantes a analizar, al igual que hicimos en el ejercicio anterior. Suba al historial el siguiente fichero único y tabulado: https://bioinfo2.ugr.es/SecuenciasDatasets/MethylationData/contaminants_list.txt
Control de calidad
- Para inspeccionar la calidad de las lecturas secuenciadas utilizaremos la herramienta FastQC al igual que usamos en el ejercicio anterior. ¿Qué diferencias observamos? ¿Interpreta los gráficos obtenidos?
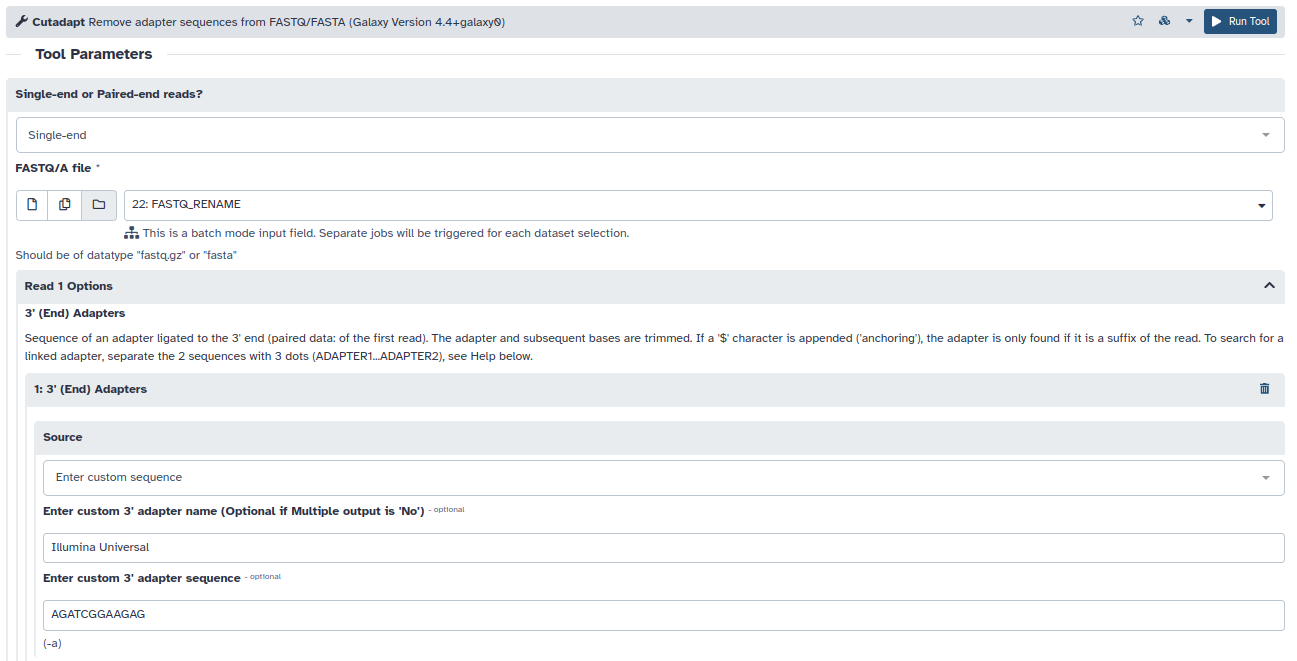
- A pesar de que los datos son buenos en general, mediante la herramienta cutadapt eliminaremos la parte de las lecturas con peor calidad e intentaremos recortar cualquier secuencia adaptadora que quede en las lecturas.
- Recortaremos los nucleótidos con baja calidad de secuenciación en el extremos 3′ de nuestras secuencias: Q<20. Quality cutoff = 20
- Identificaremos los 12 primeros nucleótidos del adaptador universal de illumina en 3′ de las lecturas recortadas por calidad: AGATCGGAAGAG. 3′ End Adapters –> Enter custom sequence
- Y descartaremos aquellas lecturas por debajo de la mitad de su longitud inicial tras los recortes. Minimum Length = 50
- Una vez obtenidos los resultados de cutadapt, podemos correr de nuevo fastQC para comparar los resultados con el resultado original.
¿Deberíamos eliminar duplicados en este experimento como hicimos en la identificación de variantes?
Alineamiento
- Durante la conversión con bisulfito las citosinas no-metiladas han sido transformadas en timinas lo que supone una reducción en la complejidad del alfabeto de ADN en algunas regiones. Como comentamos en teoría existen numerosas opciones para alinear y recuperar la información de la metilación del ADN. Sin embargo, algunas presentan un costo computacional inviable y otras pueden generar sesgos indeseados en la cuantificación final.
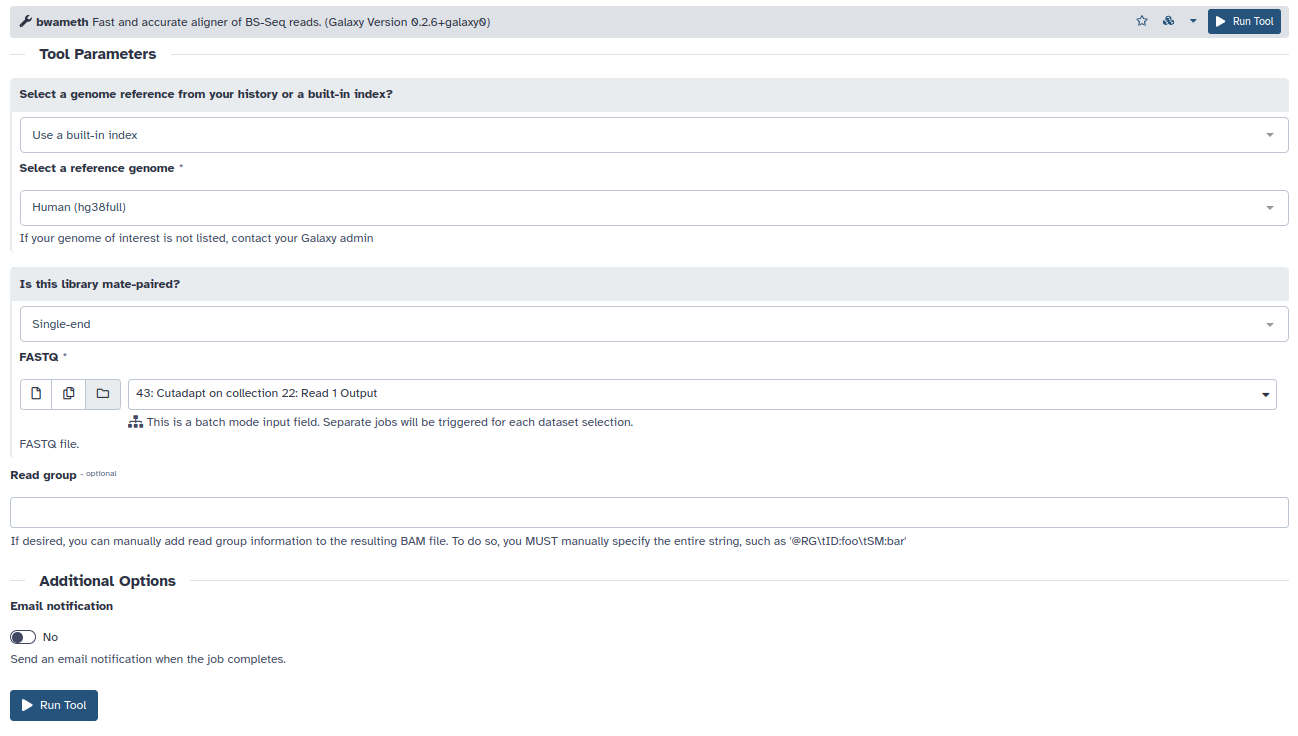
- Generalmente, la aproximación más aceptada es convertir todo el ADN a un alfabeto de 3 letras y guardar la información de los cambios, para posteriormente identificar donde se produjeron cambios o no. Para este propósito es necesario un algoritmo específico de alineamiento que lleve a cabo este proceso, nosotros utilizaremos bwameth. Seleccionaremos el genoma hg38 y nuestra colección de fastq tras recortarlos con cutadapt. Dependiendo de la carga de los servidores de galaxy este proceso puede tardar un rato.
- Una vez alineados, podemos visualizar nuestras lecturas en el buscador de UCSC. Visualize –> Display at UCSC.
¿Qué significan las variantes encontradas a lo largo de toda la región secuencia? Recordatorio: nuestra región es chr22:30000000-50000000