Cada estudiante dispone de un genoma ensamblado mediante el programa Spades
(vease Ensamblando un genoma para mas información)
Objetivos:
- Determinar la calidad del ensamblado
- Determinar la especie (cepa)
- Predecir todos los genes codificantes
- Anotar los genes predichos
- Determinar la presencia de ciertos genes que provocan resitencia a anitbiotiocos
Calidad del ensamblado
Número de contigs
Paso1: filtrar las lineas que contiene ‘>’
grep '>' /opt/bioinfo_genomas/14.fa
Paso2: contar el número de lineas
wc -l 14.fa
Paso3: juntar los dos comandos
grep '>' /opt/bioinfo_genomas/14.fa | wc -l
Número de contigs más largos que un umbral dado
contigs más largos que 1000nt
awk -v RS='>' '{ split($0,f,"_"); if (f[4] > 1000) print ">" $0; }' a14.fa
reenviar la salida a pantalla a un fichero (mediant el operados >):
awk -v RS='>' '{ split($0,f,"_"); if (f[4] > 1000) print ">" $0; }' a14.fa > a14_1000.fa
¿Que es awk?
“Dentro de las herramientas del sistema UNIX awk es equivalente a una navaja del ejercito suizo, que es útil para modificar archivos, buscar y tranformar bases de datos, generare informes simples y otras muchas cosas.”
Jesús Alberto Vidal Cortés,
Introducción en awk
desglose del comando:
| -v RS=’>’ | se lee la entrada en bloques comprendidos entre el caracter ‘>’ |
| $0 | la variable que contiene la información del bloque (String – cadena de caracteres). En este caso, el encabezado (menos la ‘>’) y la secuencia |
| split($0,f,”_”) |
NODE_244_length_1010_cov_1.952840 Cortar la cadena de caracteres en un patron dado, ‘_’ en este caso. El resultado es una lista con las subcadenas accesibles a traves de diferentes indices
|
| print “>” $0; |
print escribe una cadena de caracteres en la pantalla. En este caso, la cadena consiste en juntando ‘>’ con lo almazenado en $0 |
| > 26_1000.fa |
redirigir la salida a pantalla a un fichero |
Cuestiones
- ¿Como podemos obtener todas las longitudes en la salida?
- ¿Como podemos extraer una secuencia (scaffold) en formato fasta?
- Analizar la variación en el G+C a lo largo del contig mas largo
- ¿Podemos determinar el orden real de los contigs en el genoma?
Determinar los genes codificantes
La anotación de genes codificantes se basa en la predicción de genes en procariotas que carecen de intrones
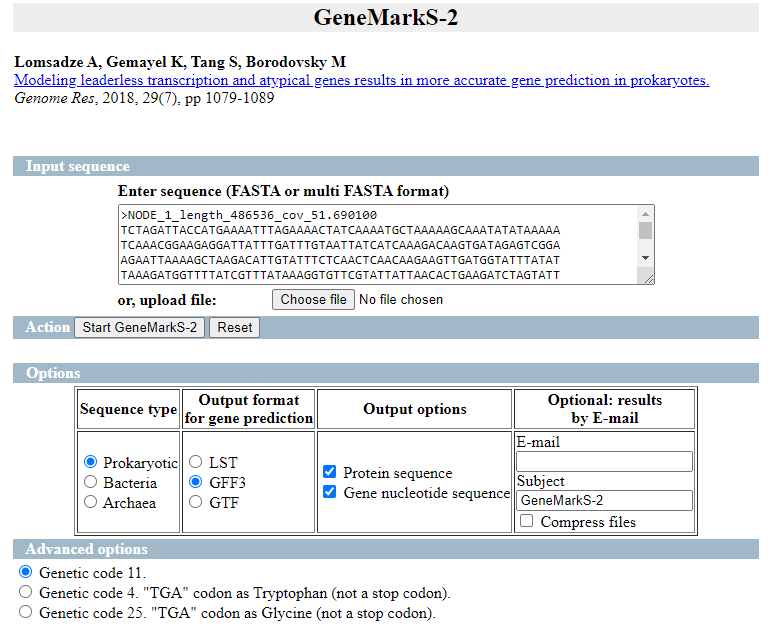
Primero usaremos GeneMarkS-2 para predecir los genes un el ensamblado mediante el fichero con contigs >= 1000nt
Marcamos las opciones como en la siguiente imagen:
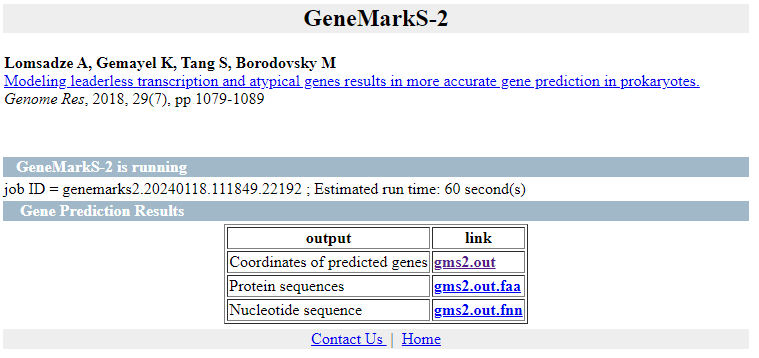
Obtenemos 3 ficheros de salida:
- la anotación de las regiones CDS en formato gff3
- Las secuencias de las regiones codificantes
- Las CDSs traducidas
Anotar los genes predichos
Existen diferentes programas:
- MicrobeAnnotator: publicación, github
- blastkoala: publicación, herramienta
- Ejemplo de salida
Usaremos el programa blastkoala que emplea Blast para asignar la secuencia del gen predicho (anónima) a una secuencia de la base de datos KEGG.
Analizar la salida del blastkoala
- Determinar el % de genes predichos asingados a una entrada de KEGG
- ¿Es posible asignar genes predichos sin entrada en KEGG mediante Blast?
Cuestiones
¿Como podemos ‘mejorar’ la tasa de genes asignados?
Solución
- Intentar usar Blast con la base de datos nr
Problema:
- Mediante el servicio web del Blast tendríamos que enviar cientos de secuencias manualmente, lo cual es inviable
![]()
Solución
Usar una versión local con una base de datos ‘customizada‘
- Detectar la especie/cepa mediant Blast
- Descargar las secuencias de la CDS y proteinas
- Generar la base de datos local
- Ejecutar una búsqueda local
Resultados:
-
Un posible genoma de referencia podría ser Staphylococcus aureus strain RJ1267
- Send to –> coding sequences –> fichero–> CP047321_RJ1267.txt
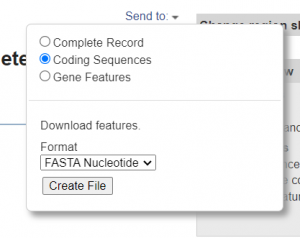
- Usar el programa de EMBOSS transeq para traducir las CDS (o la opción de guardar los CDS como ‘FASTA Protein’)
- Generar la base de datos
makeblastdb -in CP047321_RJ1267.txt -parse_seqids -title "CP047321_RJ1267" -out CP047321_RJ1267 -dbtype nucl
makeblastdb -in CP047321_RJ1267_prot.txt -parse_seqids -title "CP047321_RJ1267" -out CP047321_RJ1267 -dbtype prot
5. Ejecutar un Blast local
Suponemos que la salida del GeneMarkS-2 se llama gms2_a26.faa (secuencias de aminoacídicas) y gms2_a26.fnn
blastp -query gms2_a26.faa -db CP047321_RJ1267 -max_target_seqs 1 -outfmt 6 -evalue 1E-5 -out gms2_a26_blastp.txt
blastn -query gms2_a26.fnn -db CP047321_RJ1267 -max_target_seqs 1 -outfmt 6 -perc_identity 90 -evalue 1E-10 -out gms2_a26_blastn.txt
Véase la explicación del fichero de salida
Preguntas:
- ¿Cuantos genes predichos podemos anotar?
- Para cuantos genes predichos:
- ¿La anotación y el gen predicho tienen igual longitud?
- ¿El gen predicho alinea por encima del 90% (-qcov_hsp_perc)?
- Explorar la salida: -outfmt “6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen qcovs”
- ¿Cuantos genes no hemos podido asignar?
- ¿Como podemos lograr que en la tabla de salida salga el nombre del gen?
Obtener una lista con genes predichos no asignados
grep '>' gms2_a26.fnn | cut -f 1 -d ' ' | sed 's/>//g' > genes_predichos.txt
cut -f 1 gms2_a26_blastn.txt > genes_asignados.txt
diff -y genes_predichos.txt genes_asignados.txt
Detectar la presencia de genes de resistencia a antibioticos
Detectar genes no-codificantes
Una detección exhaustiva podemos llevar a cabo mediante Infernal (véase predicción de genes)
cmscan --rfam --cut_ga --nohmmonly --tblout mrum-genome.tblout --fmt 2 --clanin /home/biocomp/RFAM/Rfam.clanin /home/biocomp/RFAM/Rfam.cm /home/biocomp/bioinfo_genomas/26_1000.fa
Pero tambien existe la posibilidad de buscar la secuencia, pero ejemplo del gen del ARN ribosómico en el ensamblado
Otras cuestiones de interés
¿Como de buena es la predicción de genes? –> bondad predicción de genes