El objetivo de esta práctica será el de cuantificar la bondad de la predicción del programa GeneMark.
Para guiarnos, nos fijaremos en la figura 1 de la siguiente publicación: Burset M1, Guigó R. (1996) Evaluation of gene structure prediction programs.
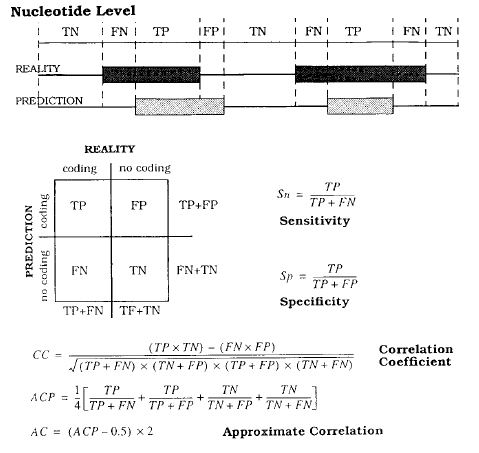
Protocolo:
1) Descargar la secuencia del genoma completo de Rickettsia massiliae que se usará para la predicción de genes. Cambiar el nombre de la secuencia a ‘CP000683’ y eliminar la descripción
2) usar el ORFfinder para detectar ORFs sobre una subsecuencia
3) Descargar las secuencias de las CDS de los genes anotados de Rickettsia massiliae. Estos datos se usarán para obtener la anotación de la secuencia con la que tenemos que comparar la predicción.
4) Predecir los genes mediante el GeneMark y guardar el resultado en formato ‘GFF’. Eliminar la última línea en blanco.
5) Eliminar el encabezado (las primeras 9 líneas) del fichero en GFF (resultado).
6) Obtener la anotación
El fichero con las secuencias de las CDS contiene para cada secuencia una anotación en formato fasta.
>lcl|CP000683.1_cds_RMA_0001_1 [gene=RMA_0001] [protein=hypothetical protein] [protein_id=ABV84350.1] [location=1..822]
El objetivo será extraer las coordenadas dadas por el tag location=1..822. Mediante una línea de comando:
cat Rickettsia_cds.fa | awk ‘{ if($1 ~ />/){ if($0 ~ /location=complement/){ split($0,f,”location=complement\\(“); split(f[2],f2,”\\.\\.”); gsub(“]”, “”,f2[2]); gsub(“\\)”, “”,f2[2]); print “CP000683\t”f2[1]-1″\t”f2[2]”\t-\t0\t-“; } else{ split($0,f,”location=”); split(f[2],f2,”\\.\\.”); gsub(“]”, “”,f2[2]); print “CP000683\t”f2[1]”\t”f2[2]”\t-\t0\t+”;} }} ‘ > rickettsia_cds_coord.txt
obtendremos una salida que consta de las coordenadas en formato bed
–desglosando el comando
escribir los encabezados
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { print $0 } }’
escribir los encabezados de las CDS de genes localizados en la hebra (-)
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=complement/ ){ print $0 } }}’
cortar la cadena de caracteres en ‘location=complement(‘; Las coordenadas estaran en el indice 2 del array f (f[2])
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=complement/ ){ split($0,f,”location=complement\\(“); print f[2] } }}’
cortar las coordenadas en ‘..’;
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=complement/ ){ split($0,f,”location=complement\\(“); split(f[2],f1,”\\.\\.”) ; print f1[1]” “f1[2] ;} }}’
eliminar ‘([‘ y escribir las coordenadas en formato BED
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=complement/ ){ split($0,f,”location=complement\\(“); split(f[2],f1,”\\.\\.”) ; gsub(“\\)]”,””,f1[2]); print “CP000683\t”f1[1]-1″\t”f1[2]”\t-\t0\t-” ;} }}’
redireccionar la salida a un fichero
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=complement/ ){ split($0,f,”location=complement\\(“); split(f[2],f1,”\\.\\.”) ; gsub(“\\)]”,””,f1[2]); print “CP000683\t”f1[1]-1″\t”f1[2]”\t-\t0\t-” ;} }}’ > annotation_-.bed
ahora para la hebra (+)
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=[0-9]/ ){ split($0,f,”location=”); print f[2] } }}’
cat sequence-2.txt | awk ‘{ if($0 ~ />/) { if($0 ~ /location=[0-9]/ ){ split($0,f,”location=”); split(f[2],f1,”\\.\\.”) ; gsub(“]”,””,f1[2]); print “CP000683\t”f1[1]-1″\t”f1[2]”\t-\t0\t+” ; } }}’ > annotation_+.txt
unir los dos ficheros
cat annotation_+.txt annotation_-.txt > annotation.txt
- Expresiones condicionales en awk
- funciones tipo ‘String’ (cadena de caracteres)
https://www.gnu.org/software/gawk/manual/html_node/String-Functions.html
- arrays en awk
http://www.delorie.com/gnu/docs/gawk/gawk_119.html
7) Usar Galaxy para comparar la predicción con la anotación (mirrors)
- Subir los datos a Galaxy: Get Data –> Upload File, especificando directamente los formatos BED (para la anotación) y GFF para la predicción.
- Convertir GFF en BED: Herramienta lápiz –> Convert Format
- Filtrar por hebra: Filter and Sort –> Filter (c6 == ‘+’ and c6 ==’-‘)
- Comparar las listas: Operate on Genomic Intervals –> Coverage